Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2015; 6(8):717-726. doi:10.7150/jca.10865 This issue Cite
Review
Perspective of Targeting Cancer-Associated Fibroblasts in Melanoma
Linli Zhou1, Kun Yang1, Thomas Andl2, R. Randall Wickett1, Yuhang Zhang1 ![]()
1. Division of Pharmaceutical Sciences, College of Pharmacy, University of Cincinnati, Cincinnati, OH 45267, USA
2. Division of Dermatology, Department of Medicine, Vanderbilt University, Nashville, TN 37232-2600, USA
Received 2014-10-21; Accepted 2015-5-4; Published 2015-6-23
Abstract
Melanoma is known as an exceptionally aggressive and treatment-resistant human cancer. Although a great deal of progress has been made in the past decade, including the development of immunotherapy using immune checkpoint inhibitors and targeted therapy using BRAF, MEK or KIT inhibitors, treatment for unresectable stage III, stage IV, and recurrent melanoma is still challenging with limited response rate, severe side effects and poor prognosis, highlighting an urgent need for discovering and designing more effective approaches to conquer melanoma. Melanoma is not only driven by malignant melanocytes, but also by the altered communication between neoplastic cells and non-malignant cell populations, including fibroblasts, endothelial and inflammatory cells, in the tumor stroma. Infiltrated and surrounding fibroblasts, also known as cancer-associated fibroblasts (CAFs), exhibit both phenotypical and physiological differences compared to normal dermal fibroblasts. They acquire properties of myofibroblasts, remodel the extracellular matrix (ECM) and architecture of the diseased tissue and secrete chemical factors, which all together promote the transformation process by encouraging tumor growth, angiogenesis, inflammation and metastasis and contribute to drug resistance. A number of in vitro and in vivo experiments have shown that stromal fibroblasts promote melanoma cell proliferation and they have been targeted to suppress tumor growth effectively. Evidently, a combination therapy co-targeting tumor cells and stromal fibroblasts may provide promising strategies to improve therapeutic outcomes and overcome treatment resistance. A significant benefit of targeting CAFs is that the approach aims to create a tumor-resistant environment that inhibits growth of melanomas carrying different genetic mutations. However, the origin of CAFs and precise mechanisms by which CAFs contribute to melanoma progression and drug resistance remain poorly understood. In this review, we discuss the origin, activation and heterogeneity of CAFs in the melanoma tumor microenvironment and examine the contributions of stromal fibroblasts at different stages of melanoma development. We also highlight the recent progression in dissecting and characterizing how local fibroblasts become reprogrammed and build a dynamic yet optimal microenvironment for tumors to develop and metastasize. In addition, we review key developments in ongoing preclinical studies and clinical applications targeting CAFs and tumor-stroma interactions for melanoma treatment.
Keywords: skin cancer, melanoma, cancer-associated fibroblasts, tumor microenvironment
Introduction
Advanced melanoma is recognized as an exceptionally aggressive and treatment-resistant human cancer. According to data from the American Cancer Society, melanoma accounts for less than 2% of skin cancer cases but a large part of skin cancer deaths. The incidence of melanoma has been rising over the past 30 years in the US. In 2014, an estimated 76,100 new cases were diagnosed and over 9,000 patients died [1], highlighting an urgent need to discover as yet unknown molecular features of melanoma in order to make fundamental therapeutic advances to conquer this malignant disease.
Emerging Approaches for Melanoma Treatment
Survival of melanoma patients largely depends on the stage of the tumor. Early stage melanoma is highly curable by a wide local excision followed by lymph node management [2]. However, treatments for patients with unresectable stage III, stage IV and recurrent melanoma have been extremely challenging without significant improvement in survival rates [3-6]. Interleukin-2 (IL-2) and interferon-alpha (IFNα)-based immunotherapy were introduced in the 1990s to treat advanced melanoma [7, 8]. In the last decade, a deeper understanding of melanoma etiology, development and metastasis has driven the rapid development of innovative agents for immunotherapy and molecularly targeted therapy. An array of immune checkpoint inhibitors has recently been approved to treat advanced melanoma. Ipilimumab (Yervoy®), a monoclonal antibody that directly binds to cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) on T cells, has the ability to reverse the CTLA-4-mediated down-regulation of T-cell activation and enhance the immune response against melanoma cells [9]. In clinical trials, patients treated with different concentration of ipilimumab showed a 5-year survival rate that varied between 16.5% and 49.5% [10]. Pembrolizumab (formerly lambrolizumab; Keytruda®) and Nivolumab (Opdivo®) block the interaction between programed cell death protein 1 (PD-1) and its two ligands, PD-L1 and PD-L2, which are highly expressed on melanoma cells, augmenting the antitumor immune response. Both drugs have shown promising clinical outcomes of reducing tumor size and increasing overall survival of patients with metastatic melanoma [11]. On September 4, 2014, the US Food and Drug Administration (FDA) approved pembrolizumab as a breakthrough therapy for treatment following other immune and molecularly targeted therapies [12]. The FDA approved Nivolumab in December 2014 as a treatment for patients who have progressed on treatment with ipilimumab [13].
Molecularly targeted therapies are drugs that specifically interfere with key molecules involved in tumor cell growth and survival. Over 60% of primary cutaneous melanomas and over 50% of metastatic melanomas harbor activating mutations in v-raf murine sarcoma viral oncogene homolog B (BRAF), mainly at residue 600 that encodes valine [14-16], resulting in the activation of mitogen-activated protein kinase (MAPK)/extracellular signal-regulated kinase (ERK) signaling without the need for extracellular stimulation [17]. Vemurafenib/PLX4032 (Zelboraf®), an inhibitor of mutated protein BRAF(V600E) from Genentech, was approved by the FDA, Health Canada and the European Commission in 2011 and 2012 as a monotherapy for metastatic melanoma [18, 19]. The relative reduction in the risk of death by vemurafenib was 63% in melanoma patients carrying BRAF mutations and the median progression-free survival is 5 to 7 months [20]. In 2013, the FDA approved two new inhibitors developed by GlaxoSmithKline, Dabrafenib (Tafinlar®) and Trametinib (Mekinist®), which showed similar outcomes to vemurafenib during phase I and II studies for treating certain patients with metastatic or unresectable melanoma carrying BRAF(V600E) or (V600K) mutations. Dabrafenib is another BRAF kinase inhibitor but appears to be more selective to the BRAF(V600E) mutation and showed a lower degree of side effects than vemurafenib [21, 22]. Trametinib acts as an inhibitor of mitogen-activated, extracellular signal-regulated kinase (MEK) and was approved in May 2013 as a single-agent treatment by the FDA for the treatment of melanoma patients with V600E or V600K mutation [23]. In January 8, 2014, the FDA approved the combination of dabrafenib and trametinib to treat patients with BRAF(V600E/K) metastatic melanoma [12].
Mutations in mast/stem cell factor growth factor receptor (SCFR), also known as tyrosine-protein kinase c-KIT or CD117, are primarily associated with acral and mucosal melanomas and a subset of cutaneous melanoma caused by chronic sun damage [24, 25]. Stem cell factor (SCF) binds to c-KIT, which is expressed on the surface of melanocytes, to activate intracellular tyrosine kinase activity and downstream multiple signaling cascades, including the RAS/ERK, phosphatidylinositol-3-OH kinase/AKT (PI3K/AKT) and Janus kinase (JAK)/signal transducer and activator of transcription (STAT) pathways, promoting cell growth and survival [26]. c-KIT gene mutations or increased c-KIT gene copy numbers abnormally activate downstream signaling processes, fostering unrestricted melanoma cell proliferation and enabling tumor cells to evade apoptosis [25, 27, 28]. There are several multicenter phase II trials for the treatment of this specific melanoma subtype using small-molecule tyrosine kinase inhibitors, including imatinib (Gleevec®), sunitinib (Sutent®) and nilotinib (Tasigna®) [29, 30].
Limitations of Immunotherapy and Molecularly Targeted Therapy in Melanoma Treatment
Although newly developed therapeutic approaches have shown promising outcomes in treating advanced melanoma, their applications obviously have limitations because of limited response rate, severe side effects and poor prognosis. Especially, targeted therapies using BRAF, MEK or KIT inhibitors can only deal with a particular subtype of melanoma carrying specific mutations. Furthermore, melanoma cells frequently implement a number of molecular and cellular mechanisms to become resistant to these drugs, including higher mutation rate, alteration of membrane drug transporters, increased DNA repair and dysfunction of apoptosis [31, 32]. Heterogeneity within melanomas has been revealed as another possible mechanism behind tumor resistance to therapeutic treatment [33].
Drug resistance in melanoma can be either pre-existent (intrinsic resistance) or induced by treatment (acquired resistance) [34, 35]. Intrinsic resistance causes poor response to the anti-melanoma drugs. For example, clinical studies reported only about 10% of patients responding to ipilimumab [36]. A research report published by Cha et al. in 2014 showed that patients who maintained stable pre-existing T cell clonotypes had a good response to ipilimumab and improved overall survival [37]. Acquired resistance occurred in melanomas treated with BRAF inhibitors such as vemurafenib [38]. One primary cause is the reactivation of the MAPK and PI3K/AKT signaling pathways [39], which can be induced by secondary mutations in genes downstream of BRAF, such as those encoding neuroblastoma RAS viral (v-ras) oncogene homolog (NRAS) [40] or MEK1 [41]. Hepatocyte growth factor (HGF) secreted by stromal fibroblasts has been revealed as another cause that allows melanoma cells to bypass BRAF inhibition and maintain abnormal cell cycle progression [42] .
Another drawback limiting the application of newly developed drugs is the occurrence of severe side effects [43, 44]. Treatment with IFNα causes depression, flu-like symptoms, fatigue, anorexia and neuropsychiatric symptoms. IL-2 treatment can induce vascular leak syndrome with symptoms of hypotension and oliguria [45]. Autoimmune-mediated side effects are reported among patients treated with ipilimumab, including mild cutaneous disorders such as skin rash, life-threatening toxic epidermal necrolysis and colitis. Although many patients showed good tolerance to vemurafenib, cutaneous toxicities still developed, including verrucous papilloma, hand-foot skin reaction, rash, photosensitivity, panniculitis, and especially keratoacanthomas and squamous cell carcinomas. Interestingly, it has been reported that these side effects correlated with increasing positive response of metastatic melanoma to vemurafenib [46, 47].
Alternative Strategies to Overcome Tumor Cell Heterogeneity and Therapy Resistance: Targeting Cancer-Associated Fibroblasts (CAFs)
Solid tumors are not monolithic tumor cell-only masses but also contain a wide range of non-cancer stromal cells including endothelial cells, fibroblasts, mesenchymal stem cells and immune cells [48]. Infiltrated and surrounding fibroblasts in the tumor microenvironment, termed CAFs, have the ability to synthesize, deposit and remodel the ECM and produce cytokines and growth factors, which all together promote the transformation process by encouraging tumor growth, angiogenesis and inflammation and contribute to drug resistance [49]. The presence of a larger number of CAFs in the tumor stroma has been shown to be associated with an increased risk of metastasis and a poor clinical prognosis in breast cancer, lung cancer, and pancreatic cancer [50, 51]. Several co-culture experiments demonstrated that CAFs stimulated melanoma, pancreatic and prostatic cancer cell growth in vitro [52-54]. A number of in vivo experiments provided evidence that CAFs promoted melanoma cell proliferation and thus they were targeted to suppress tumor growth [54-58].
CAFs are believed to be heterogeneous with similar properties to the activated myofibroblasts found under inflammatory conditions or during wound healing, and normally express α-smooth muscle actin (α-SMA, also called ACTA2) [59]. However, α-SMA alone cannot be used to identify all CAFs. Several other markers such as, fibroblast specific protein 1 (FSP-1, also called S100A4), fibroblast activation protein-alpha (FAP-α, also known as FAP) and platelet-derived growth factor receptor (PDGFR) have also been used to identify, isolate and characterize CAFs [51, 60].
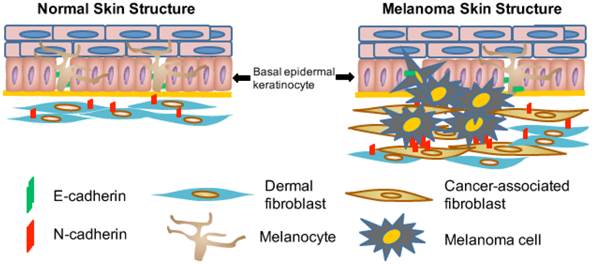
There are several important sources for the formation of CAFs in the tumor stroma, including: (1) transdifferentiation of bone marrow mesenchymal cells into CAFs; (2) epithelial-mesenchymal transition (EMT) or endothelial-to-mesenchymal transition (EndMT) of resident epithelial or endothelial cells within the tumor stroma into CAFs; and (3) recruitment and activation of resident fibroblasts [61, 62]. As cutaneous melanoma begins predominantly at the junction of the epidermis and the dermis (Fig. 1), malignant tumor cells interact actively and extensively with dermal fibroblasts [63], which undoubtedly serve as a major source for CAFs and play an important role in melanoma development and metastasis.
Illustration of interactions among melanocytes, keratinocytes and fibroblasts in normal skin and during melanoma development. Normal melanocytes reside in the epidermis and close to the basement membrane forming an 'Epidermal Melanin Unit' that contains one melanocyte and up to thirty-six keratinocytes. Malignant melanoma cells proliferate, penetrate the basement membrane (thick yellow line), and invade the underlying dermis. Melanocytes are adherent to keratinocytes due to the E-cadherin expression (indicated by green bars). During the process of melanoma invasion and metastasis, the switch from E-cadherin to N-cadherin expression in melanoma cells allows them to bind to N-cadherin-expressing fibroblasts (indicated by red bars), which subsequently helps these melanoma cells move away from keratinocytes and invade the underlying dermis before they can reach blood and lymphatic vessels [86].
Inhibitory Functions by Dermal Fibroblasts to Suppress Tumor Onset
In contrast to CAFs, it appears that normal dermal fibroblasts suppress growth and progression of pre-malignant lesions at early stages of tumor development (Fig. 2) [64]. In one study, dermal fibroblasts showed a repressive influence on tumor cells derived from primary melanoma lesions using an in vitro co-culture model [54]. However, mechanisms that normal fibroblasts employ to defend against tumor development are still under investigation. Several research papers reported that the possible cause of this inhibitory effect by normal dermal fibroblasts is based on their abilities to mobilize immune cells by secreting cytokines such as interferon gamma (IFNγ), interleukin-6 (IL-6) and tumor necrosis factor-alpha (TNF-α) [65, 66]. Madar et al. reported that dermal fibroblasts expressed high levels of tumor inhibitory proteins, such as whey acidic protein four-disulfide core domain 1 (WFDC1), which suppressed tumor cell growth [67, 68].
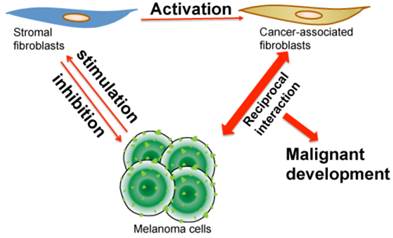
Illustration of reciprocal interaction and co-evolution among stromal fibroblasts, cancer-associated fibroblasts and melanoma cells. When melanoma first initiates, the quiescent fibroblasts in stroma create a barrier and produce chemical factors, which altogether inhibit melanoma cell growth. During the development of melanoma, melanoma cells secret stimulatory molecules, which induce epigenetic, cellular and biochemical changes in stromal fibroblasts that consequently become CAFs. After activation, CAFs subsequently interact with melanoma cells in a reciprocal way that builds a dynamic yet optimal microenvironment for melanoma to progress, metastasize and resist drug treatment.
Another possible reason is the physical barrier formed by dermal fibroblasts that blocks melanoma cells to migrate and invade the surrounding tissues. The ability of malignant melanoma cells to penetrate dermal tissue requires changes in several distinct cellular functions including adhesion, motility, detachment, and ECM proteolysis. Members of the matrix metalloproteinase (MMP) gene family have been implicated in the remodeling of the ECM required for tumor invasion and metastasis. Normal fibroblasts continually regulate and restrain the change of the ECM in healthy tissue by strictly controlling MMP1, MMP2, MMP9, and MMP13, and membrane-type matrix metalloprotineases (MT-MMPs), thereby preventing the degradation of basement membrane and consequently blocking the potential invasion and metastasis of tumor cells [69].
ECM proteins and secreted factors expressed by CAFs may also exert tumor suppressive effects at the early stages of tumor progression. Hyaluronan is a large polysaccharide usually consisting of 2,000-25,000 disaccharides [70]. It has been reported that the majority of hyaluronan in the ECM was synthesized by CAFs and promoted tumor invasiveness [71, 72]. However, hyaluronidase found in stromal tissues cleaved hyaluronan into polymers of various sizes, which showed different biological activities. Small size hyaluronan oligomers suppressed the MAPK and PI3K/AKT pathways in multidrug-resistant tumor cells and sensitized cancer cells to a variety of chemotherapeutic drugs [73-75]. IL-6 was reported to be a fibroblast-derived growth inhibitor of early stage human melanoma cells [65]. Transforming growth factor-beta (TGF-β), for which fibroblasts are an important source, is known to suppress melanocyte and early stage melanoma cell cycle progression [60].
Stimulation and Transformation of Stromal Fibroblasts to CAFs
CAFs exhibit both phenotypical and epigenetic differences from normal dermal fibroblasts, which appear to be quiescent under normal healthy conditions and function to maintain tissue homeostasis [59]. CAFs isolated from breast cancer tissue showed higher motility and invasion than normal fibroblasts [76]. Although both CAFs and normal fibroblasts express fibroblast-specific markers, such as fibronectin and vimentin, molecules that are normally considered to be activation markers, including α-SMA, S100A4, and FAP-α, are only expressed in CAFs [77]. Furthermore, expression of genes that regulate cell proliferation, cell death and cell adhesion was found to be highly up-regulated in CAFs derived from malignant tumors as compared with normal fibroblasts from breast and prostate tissue [78-80].
How dermal fibroblasts are stimulated to enter a continuous state of irreversible activation and establish an environment conducive for melanoma development remains poorly understood. As shown in Fig. 2, possible drivers are stimulatory factors that are secreted by tumor cells during carcinoma development [81]. By using a 3-dimensional (3D) collagen matrix, Anderson and his collaborators showed that chemical factors secreted by melanoma cells induced fibroblasts to migrate toward, surround, and then infiltrate the tumor mass. These signals also stimulate fibroblasts to produce ECM proteins and a pool of pro-tumorigenic soluble factors [82]. They confirmed that platelet-derived growth factor (PDGF) and basic fibroblast growth factor (bFGF) were present in melanoma cell-conditioned medium and could stimulate an up to 6-fold increase in the production of glycosaminoglycan (GAG) from fibroblasts [83]. In addition, TGF-β is abundant in melanoma lesions and can stimulate normal fibroblasts to become CAFs [84].
CAFs Promote Tumor Growth and Invasion by Direct Cell-Cell Interaction
Following stimulation, transdifferentiated CAFs can promote tumor growth and invasion through direct cell-cell contact. CAFs were found to have a better ability to induce EMT and motility of non-small-cell-lung cancer cells when the two cell populations were cultured together permitting direct cell-cell contacts than when they were co-cultured indirectly across a trans-well membrane without direct physical contact [85]. As shown in Fig. 1, melanocytes are surrounded by keratinocytes in the epidermis of normal skin. Growth, dendricity, and expression of melanoma-associated cell surface molecules in melanocytes are restricted since melanocytes are adherent to keratinocytes partly because of E-cadherin expression and resulting direct interaction [86, 87]. As a tumor progresses, the switch from E-cadherin to N-cadherin expression controlled by PTEN/PI3K signaling allows mutant melanocytes to bind to N-cadherin-expressing fibroblasts, and subsequently helps them to move away from keratinocytes and invade the underlying dermis [88-90].
CD44 is a multifunctional glycoprotein that is abundantly expressed in CAFs and contributes to the interaction between CAFs and tumor cells [91]. CD44 has been shown to increase the motility and tumorigenicity of murine mammary and human head and neck cancer cells [92, 93]. In one study conducted by Kinugasa et al. to investigate the effect of CD44 on tumorigenicity of HT29 cancer stem cells (CSCs), CSCs mixed with wild-type CAFs formed 2-fold more spheroids than the CSCs did alone, while CSCs cultured with CD44-deficient CAFs or in CAF-conditional medium did not show increased formation of spheroids. This observation clearly suggested that CD44-mediated direct interaction between CAFs and carcinoma cells contributes to stemness, tumorigenicity and drug resistance of CSCs [91].
CAFs Modulate the ECM to Promote Tumor Growth and Invasion
CAFs produce a variety of ECM proteins such as laminins, fibronectin, tenascin C (TN-C), and fibrillar collagens, which are the structural components that make up connective tissue and contribute to the dense fibrous nature of solid tumors [94]. As discussed above, the hyaluronan-rich matrix facilitates tumor growth, invasion and angiogenesis. Remodeling of the ECM by MMPs is one crucial step in the formation of a favorable tumor microenvironment, allowing cancer cells to escape the primary tumor site and cross tissue boundaries [60]. In vitro evidence showed MMP1, MMP2, MMP13 and MT1-MMP (MMP14) secreted by surrounding CAFs influenced the motility and invasiveness of melanoma cells [95-99]. In primary and metastatic melanomas up-regulated expression of FAP-α in the reactive mesenchyme was also observed [100]. As an active serine protease, FAP-α has both collagenolytic and dipeptidyl peptidase activities capable of degrading type I collagen, thus enhancing ECM remodeling and facilitating tumor cell growth and migration [101, 102].
CAFs Secrete Various Factors to Promote Melanoma Growth and Invasion
CAFs have the ability to synthesize and secrete a large variety of growth factors and cytokines to support malignant tumor cell growth, invasion and metastasis [62, 103]. One possible mechanism suggested by Comito et al. and Taddei et al. is that hypoxia-induced oxidative stress in melanoma and chemical factors secreted by melanoma cells stimulate CAFs to secrete cytokines and growth factors such as vascular endothelial growth factor (VEGF), stromal derived factor-1 (SDF-1 or CXCL12) and IL-6 favoring melanoma chemotaxis and invasion [104, 105].
Cytokines from CAFs
CAFs are believed to be an important source of chemokines, including C-C motif chemokine ligand (CCL) and C-X-C motif chemokine ligand (CXCL). CCL and CXCL interact with the chemokine receptors to alter the gene expression profile of CAFs and melanoma cells, regulate their interaction and stimulate melanoma progression and metastasis [106]. Increased production of a number of chemokines including CCL2, CXCL1, CXCL2, CXCL8 (IL-8) by CAFs was observed when co-cultured with tumor cells from breast cancer, pancreatic cancer, oral cancer, and melanoma [107]. Augstern et al. identify CXCL14 expressed by CAFs as a novel paracrine stimulator of prostate cancer cell growth and migration in vitro and in vivo [108]. Expression of inflammatory chemokine CXCL8, interleukin-1 (IL-1) and CCL2 was up-regulated in CAFs when co-cultured with melanoma cells facilitating angiogenesis and tumor metastasis [107].
An interesting example of crosstalk between melanoma cells and the tumor microenvironment is the CXC chemokine receptor-4 (CXCR4)/CXCL12 axis. CXCR4 is expressed on the surface of melanoma cells and has been found to contribute to cancer metastasis, which suggests that CXCR4 expression could be a useful prognostic marker for malignant melanoma [109, 110]. CXCL12 released by CAFs in the tumor microenvironment stimulated the migration and metastasis of melanoma cells to the distal metastatic sites through interaction with CXCR4 expressed on tumor cells. CXCL12 is also able to induce angiogenesis by recruiting endothelial cells into the tumor stroma [109, 111, 112]. Hwang and coworkers have shown that CXCR4 played a role in the migration of melanoma cells from the blood to distant organs including the lung. Blockade of CXCL12-CXCR4 interaction by AMD11070, a small-molecule inhibitor of CXCR4, inhibited the migration of melanoma cells [113]. Takekoshi et al recently reported that a dimeric variant of CXCL12 (CXCL122) effectively inhibited lung metastasis of CXCR4-expressing B16/F10 melanoma cells [114].
IL-1 has been shown to activate the nuclear factor NF-κB pathway, which in turn can induce CXCL1 and CXCL2 expression in both melanoma cells and CAFs, leading to tumor progression [115, 116]. IL-6 is a fibroblast-derived factor and plays an inhibitory role in early stage melanoma but not advanced melanoma [65]. Terai et al. reported that IL-6 induced IL-10 production in melanoma cells, and subsequently IL-10 negatively regulated the immune response and helped melanoma cells escape immune surveillance [117]. Na et al. demonstrated that IL-6 activated the STAT3 signaling pathway promoting metastasis of melanoma cells [118].
Growth factors from CAFs
Growth factors such as HGF and TGF-β from CAFs play a critical role in tumor progression. TGF-β expressed by CAFs functions as a paracrine factor to drive melanoma growth and metastasis in advanced melanoma [119, 120]. HGF secreted by CAFs stimulates the proliferation of melanoma cells mediated by tyrosyl-phosphorylation of MET and several other proteins, including MAPK and ERK2 [121]. Gaggioli et al. reported that HGF also induced fibronectin expression and associated matrix assembly, which subsequently promoted melanoma metastasis [122]. Koefinger et al. proposed that the E-cadherin to N-cadherin switch in melanoma cells is mediated by HGF through stage-specific changes in expression levels of three transcription factors, SNAI1, SNAI2, and TWIST1, which were known to repress E-cadherin expression [123, 124]. VEGFs are major growth factors secreted by CAFs that support continuous growth of tumors and have a wide range of effects on stromal cells [103, 125, 126].
CAFs Contribute to Tumor Drug Resistance
Stromal fibroblasts could potentially contribute to drug resistance in tumor cells by various mechanisms including: (1) form a compact and well-organized matrix structure to prevent therapeutic drugs from reaching melanoma cells in the core of melanoma tissues [82]; (2) alter tumor cell sensitivity to apoptosis induced by anti-cancer drugs through cell-ECM interaction; (3) release soluble growth factors and chemokines that promote survival and growth of tumor cells; and (4) interact directly with tumor cells to induce epigenetic and phenotypical changes of tumor cells and activate tumor-survival signaling [127, 128]. The involvement of CAFs in drug resistance indicates that they could be promising therapeutic targets to increase the sensitivity of melanoma cells to anti-tumor agents for combined melanoma therapy.
Heterogeneity of CAFs leads to gradients of nutrition, hypoxia and acidity in the tumor mass, all of which can reduce the sensitivity of tumor cells to drug treatment [129]. A hypoxic gradient could induce oxidative stress in the tumor microenvironment and changes of gene expression in tumor cells so that tumor cells could escape from drug-induced senescence and apoptosis [128]. Oxidative stress could also arrest cells in the G1 phase of the cell cycle, which helps them to escape conventional chemotherapeutic drugs, for example, dacarbazine and temozolomide, as they usually target the rapidly dividing cells [130].
Recent data have revealed the contribution of CAF-derived molecules to drug resistance of tumor cells. Misra et al. reported that increased hyaluronan production by CAFs enhanced resistance of drug-sensitive human breast cancer cells [74]. Type I collagen from CAFs leads to decreased chemotherapeutic drug uptake in breast and colon carcinoma cells [131]. In two recent studies of melanoma drug resistance to chemotherapeutic agents, Zigrino et al. and Tiago et al. confirmed that CAFs assist melanoma metastasis and drug resistance by increasing the expression of IL-6, IL-8, MMP1, 2 and 9 [132, 133]. Studies done by Straussman et al. found that HGF-dependent stroma-mediated resistance is the possible mechanism by which melanoma cells harboring BRAF mutations evade BRAF inhibitor toxicity. HGF secreted by CAFs results in activation of the HGF receptor MET and downstream MAPK and PI3K/AKT signaling pathways, which are responsible for the immediate resistance of BRAF-mutant melanoma to BRAF inhibitors [42].
Targeting CAFs to Treat Melanoma
As CAFs play an important role in tumor development and are more genetically stable than tumor cells, CAFs have been emerging as a promising therapeutic target for melanoma treatment [131]. One major issue to consider when designing therapeutic approaches targeting CAFs or their interaction with melanoma cells is the status of the tumor and the biochemical, cellular and molecular features of the tumor microenvironment. As discussed above, molecules that are associated with CAFs or involved in tumor-stroma interaction may function as tumor promoters at one stage but become inhibitors in a different microenvironmental setting or at different tumor stages. Careful evaluation of melanoma is a prerequisite for targeting selected molecules in CAFs. Obvious examples are hyaluronan and TGF-β that we discussed previously. Therefore, an in-depth understanding of the dynamics of tumor-stroma interactions that promote melanoma cell survival, growth and invasion is critical and indispensable in the success of CAF-targeting treatment strategies. Development of new genetic, molecular and cellular tools will undoubtedly accelerate this process.
Agents targeting FAP-α, CXCR4/CXCL12, HGF, PDGF, TGF-β and hyaluronan signaling in CAFs have been under preclinical study or clinical trials for treating breast cancer, chronic myelogenous leukemia, gastrointestinal stromal tumor and melanoma [134, 135]. The acting mechanisms of these novel drugs are to either directly inhibit fibroblast growth or target signaling pathways involved in CAF-tumor cell interactions [134, 136]. Ablation of Notch1 in mouse embryonic fibroblasts sped up their growth and motility rate [137]. Further studies demonstrated that activation of the Notch1 signaling pathway in CAFs inhibited melanoma cell growth in culture and in a xenograft mouse model of human melanoma [56]. Lee et al. showed that B16/F10 melanoma development in mice was effectively suppressed after being vaccinated against FAP-α (mainly expressed on CAFs) [138]. In addition, imatinib, the tyrosine kinase inhibitor, suppressed cervical tumor growth and invasion in mice by blocking PDGFR signaling on CAFs [139].
Conclusion
Treatment options for patients with metastatic melanoma have expanded considerably in recent years but so far have failed to improve the overall survival rate [140]. This highlights the urgent need to discover and design novel and effective therapeutic approaches. The greatest benefit of targeting CAFs may be the fact that it could be applied to patients carrying different genetic mutations and help to overcome drug resistance due to melanoma heterogeneity and genomic instability.
Although exciting progress has been made in our understanding of CAFs and their reciprocal interactions with melanoma cells, the detailed mechanisms underlying these interactions remain elusive. For example, are dermal fibroblasts the only source of CAFs in melanoma? What is the origin of CAFs at the metastatic site? How many subtypes of CAFs are involved in melanoma development and metastasis? What is the function of each subtype? Therefore, further investigations of molecular mechanisms by which stromal fibroblasts are reprogrammed to acquire tumor-promoting properties, remodel the tumor microenvironment and support tumorigenicity and drug resistance will certainly advance our understanding of melanoma biology and provide new ideas to design better therapeutic strategies for this fatal disease.
Competing Interests
The authors have declared that no competing interest exists.
References
1. American Cancer Society. What are the key statistics about melanoma skin cancer?. http://www.cancer.org/cancer/skincancer-melanoma/detailedguide/melanoma-skin-cancer-key-statistics:
2. Faries MB, Ariyan S. Current surgical treatment in melanoma. Current problems in cancer. 2011;35:173-84
3. Dummer R, Hauschild A, Guggenheim M, Keilholz U, Pentheroudakis G. Cutaneous melanoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Annals of oncology: official journal of the European Society for Medical Oncology / ESMO. 2012;23(Suppl 7):86-91
4. Guadagnolo BA, Prieto V, Weber R, Ross MI, Zagars GK. The role of adjuvant radiotherapy in the local management of desmoplastic melanoma. Cancer. 2014;120:1361-8
5. Forschner A, Heinrich V, Pflugfelder A, Meier F, Garbe C. The role of radiotherapy in the overall treatment of melanoma. Clinics in dermatology. 2013;31:282-9
6. Garbe C, Eigentler TK, Keilholz U, Hauschild A, Kirkwood JM. Systematic review of medical treatment in melanoma: current status and future prospects. The oncologist. 2011;16:5-24
7. Atzpodien J, Neuber K, Kamanabrou D, Fluck M, Brocker EB, Neumann C. et al. Combination chemotherapy with or without s.c. IL-2 and IFN-alpha: results of a prospectively randomized trial of the Cooperative Advanced Malignant Melanoma Chemoimmunotherapy Group (ACIMM). British journal of cancer. 2002;86:179-84
8. Eggermont AM. The role interferon-alpha in malignant melanoma remains to be defined. Eur J Cancer. 2001;37:2147-53
9. Ipilimumab. FDA. http://www.fda.gov/aboutfda/centersoffices/officeofmedicalproductsandtobacco/cder/ucm248478.htm
10. Gogas H, Polyzos A, Kirkwood J. Immunotherapy for advanced melanoma: fulfilling the promise. Cancer treatment reviews. 2013;39:879-85
11. O'Sullivan Coyne G, Madan RA, Gulley JL. Nivolumab: promising survival signal coupled with limited toxicity raises expectations. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2014;32:986-8
12. Trametinib and Dabrafenib. FDA. http://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm381451.htm
13. Nivolumab. FDA. http://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm427807.htm
14. Maldonado JL, Fridlyand J, Patel H, Jain AN, Busam K, Kageshita T. et al. Determinants of BRAF mutations in primary melanomas. Journal of the National Cancer Institute. 2003;95:1878-90
15. Pavey S, Johansson P, Packer L, Taylor J, Stark M, Pollock PM. et al. Microarray expression profiling in melanoma reveals a BRAF mutation signature. Oncogene. 2004;23:4060-7
16. Libra M, Malaponte G, Navolanic PM, Gangemi P, Bevelacqua V, Proietti L. et al. Analysis of BRAF mutation in primary and metastatic melanoma. Cell Cycle. 2005;4:1382-4
17. Hertzman Johansson C, Egyhazi Brage S. BRAF inhibitors in cancer therapy. Pharmacology & therapeutics. 2014;142:176-82
18. Vemurafenib. FDA. http://www.fda.gov/aboutfda/centersoffices/officeofmedicalproductsandtobacco/cder/ucm268301.htm
19. Vemurafenib. Canada H. http://www.hc-sc.gc.ca/dhp-mps/prodpharma/sbd-smd/drug-med/sbd_smd_2012_zelboraf_148693-eng.php
20. Hartsough EJ, Basile KJ, Aplin AE. Beneficial effects of RAF inhibitor in mutant BRAF splice variant-expressing melanoma. Molecular cancer research: MCR. 2014;12:795-802
21. Luke JJ, Hodi FS. Ipilimumab, vemurafenib, dabrafenib, and trametinib: synergistic competitors in the clinical management of BRAF mutant malignant melanoma. The oncologist. 2013;18:717-25
22. Dabrafenib. FDA. http://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm354477.htm
23. Trametinib. FDA. http://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm354478.htm
24. Garrido MC, Bastian BC. KIT as a Therapeutic Target in Melanoma. J Invest Dermatol. 2010;130:20-7
25. Curtin JA, Busam K, Pinkel D, Bastian BC. Somatic activation of KIT in distinct subtypes of melanoma. Journal of Clinical Oncology. 2006;24:4340-6
26. Kit. Lovly C, Sosman J, Pao W. http://www.mycancergenome.org/content/disease/melanoma/kit/?tab=0
27. Satzger I, Schaefer T, Kuettler U, Broecker V, Voelker B, Ostertag H. et al. Analysis of c-KIT expression and KIT gene mutation in human mucosal melanomas. British journal of cancer. 2008;99:2065-9
28. Smalley KSM, Sondak VK, Weber JS. c-KIT signaling as the driving oncogenic event in sub-groups of melanomas. Histology and histopathology. 2009;24:643-50
29. Flaherty KT, Hodi FS, Bastian BC. Mutation-driven drug development in melanoma. Curr Opin Oncol. 2010;22:178-83
30. Minor DR, Kashani-Sabet M, Garrido M, O'Day SJ, Hamid O, Bastian BC. Sunitinib therapy for melanoma patients with KIT mutations. Clinical cancer research: an official journal of the American Association for Cancer Research. 2012;18:1457-63
31. Grossman D, Altieri DC. Drug resistance in melanoma: mechanisms, apoptosis, and new potential therapeutic targets. Cancer metastasis reviews. 2001;20:3-11
32. Helmbach H, Rossmann E, Kern MA, Schadendorf D. Drug-resistance in human melanoma. International journal of cancer Journal international du cancer. 2001;93:617-22
33. Somasundaram R, Villanueva J, Herlyn M. Chapter Eleven - Intratumoral Heterogeneity as a Therapy Resistance Mechanism: Role of Melanoma Subpopulations. In: (ed.) Keiran SMS. Advances in Pharmacology. Academic Press. 2012:335-59
34. Gottesman MM. Mechanisms of cancer drug resistance. Annual review of medicine. 2002;53:615-27
35. Lippert TH, Ruoff HJ, Volm M. Intrinsic and acquired drug resistance in malignant tumors. The main reason for therapeutic failure. Arzneimittel-Forschung. 2008;58:261-4
36. Chmielowski B. Ipilimumab: A First-in-Class T-Cell Potentiator for Metastatic Melanoma. Journal of Skin Cancer. 2013;2013:8
37. Cha E, Klinger M, Hou Y, Cummings C, Ribas A, Faham M. et al. Improved survival with T cell clonotype stability after anti-CTLA-4 treatment in cancer patients. Science translational medicine. 2014;6:238-70
38. Das Thakur M, Salangsang F, Landman AS, Sellers WR, Pryer NK, Levesque MP. et al. Modelling vemurafenib resistance in melanoma reveals a strategy to forestall drug resistance. Nature. 2013;494:251-5
39. Shi H, Hugo W, Kong X, Hong A, Koya RC, Moriceau G. et al. Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer discovery. 2014;4:80-93
40. Yadav V, Zhang X, Liu J, Estrem S, Li S, Gong XQ. et al. Reactivation of mitogen-activated protein kinase (MAPK) pathway by FGF receptor 3 (FGFR3)/Ras mediates resistance to vemurafenib in human B-RAF V600E mutant melanoma. The Journal of biological chemistry. 2012;287:28087-98
41. Rebecca VW, Alicea GM, Paraiso KH, Lawrence H, Gibney GT, Smalley KS. Vertical inhibition of the MAPK pathway enhances therapeutic responses in NRAS-mutant melanoma. Pigment Cell Melanoma Res. 2014;27:1154-8
42. Straussman R, Morikawa T, Shee K, Barzily-Rokni M, Qian ZR, Du J. et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature. 2012;487:500-4
43. Ma C, Armstrong AW. Severe adverse events from the treatment of advanced melanoma: a systematic review of severe side effects associated with ipilimumab, vemurafenib, interferon alfa-2b, dacarbazine and interleukin-2. The Journal of dermatological treatment. 2014;25:401-8
44. Ascierto PA, Bastholt L, Hersey P, Cinat G, Eggermont AM, Hauschild A. et al. Side Effects and Toxicities of Targeted Therapies in Stage IV Melanoma. American journal of therapeutics. 2015;22:44-53
45. Hauschild A, Gogas H, Tarhini A, Middleton MR, Testori A, Dreno B. et al. Practical guidelines for the management of interferon-alpha-2b side effects in patients receiving adjuvant treatment for melanoma: expert opinion. Cancer. 2008;112:982-94
46. Chon SY, Sambrano BL, Geddes ER. Vemurafenib-related cutaneous side effects ameliorated by acitretin. Journal of drugs in dermatology. 2014;13:586-8
47. Boussemart L, Routier E, Mateus C, Opletalova K, Sebille G, Kamsu-Kom N. et al. Prospective study of cutaneous side-effects associated with the BRAF inhibitor vemurafenib: a study of 42 patients. Ann Oncol. 2013;24:1691-7
48. Alphonso A, Alahari SK. Stromal cells and integrins: conforming to the needs of the tumor microenvironment. Neoplasia. 2009;11:1264-71
49. Micke P, Ostman A. Exploring the tumour environment: cancer-associated fibroblasts as targets in cancer therapy. Expert opinion on therapeutic targets. 2005;9:1217-33
50. Orimo A, Gupta PB, Sgroi DC, Arenzana-Seisdedos F, Delaunay T, Naeem R. et al. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell. 2005;121:335-48
51. Rasanen K, Vaheri A. Activation of fibroblasts in cancer stroma. Experimental cell research. 2010;316:2713-22
52. Hwang RF, Moore T, Arumugam T, Ramachandran V, Amos KD, Rivera A. et al. Cancer-Associated Stromal Fibroblasts Promote Pancreatic Tumor Progression. Cancer Res. 2008;68:918-26
53. Olumi AF, Grossfeld GD, Hayward SW, Carroll PR, Tlsty TD, Cunha GR. Carcinoma-associated Fibroblasts Direct Tumor Progression of Initiated Human Prostatic Epithelium. Cancer Res. 1999;59:5002-11
54. Cornil I, Theodorescu D, Man S, Herlyn M, Jambrosic J, Kerbel RS. Fibroblast cell interactions with human melanoma cells affect tumor cell growth as a function of tumor progression. Proceedings of the National Academy of Sciences of the United States of America. 1991;88:6028-32
55. Santos AM, Jung J, Aziz N, Kissil JL, Pure E. Targeting fibroblast activation protein inhibits tumor stromagenesis and growth in mice. The Journal of clinical investigation. 2009;119:3613-25
56. Shao H, Cai L, Grichnik JM, Livingstone AS, Velazquez OC, Liu ZJ. Activation of Notch1 signaling in stromal fibroblasts inhibits melanoma growth by upregulating WISP-1. Oncogene. 2011;30:4316-26
57. Berking C, Takemoto R, Schaider H, Showe L, Satyamoorthy K, Robbins P. et al. Transforming growth factor-beta1 increases survival of human melanoma through stroma remodeling. Cancer Res. 2001;61:8306-16
58. Coucke P, Baramova E, Leprince P, Depauwgillet M, Foidart J, Bassleer R. Metalloproteinases and serine proteases activities in mixed spheroids of mouse b16 melanoma-cells and fibroblasts. International journal of oncology. 1994;5:1125-30
59. Augsten M. Cancer-associated fibroblasts as another polarized cell type of the tumor microenvironment. Frontiers in oncology. 2014;4:63
60. Kalluri R, Zeisberg M. Fibroblasts in cancer. Nature reviews Cancer. 2006;6:392-401
61. Anderberg C, Pietras K. On the origin of cancer-associated fibroblasts. Cell Cycle. 2009;8:1461-2
62. Shimoda M, Mellody KT, Orimo A. Carcinoma-associated fibroblasts are a rate-limiting determinant for tumour progression. Seminars in cell & developmental biology. 2010;21:19-25
63. Cirri P, Chiarugi P. Cancer associated fibroblasts: the dark side of the coin. American journal of cancer research. 2011;1:482-97
64. Bissell MJ, Hines WC. Why don't we get more cancer? A proposed role of the microenvironment in restraining cancer progression. Nat Med. 2011;17:320-9
65. Lu C, Vickers MF, Kerbel RS. Interleukin 6: a fibroblast-derived growth inhibitor of human melanoma cells from early but not advanced stages of tumor progression. Proceedings of the National Academy of Sciences of the United States of America. 1992;89:9215-9
66. Cheng N, Bhowmick NA, Chytil A, Gorksa AE, Brown KA, Muraoka R. et al. Loss of TGF-beta type II receptor in fibroblasts promotes mammary carcinoma growth and invasion through upregulation of TGF-alpha-, MSP- and HGF-mediated signaling networks. Oncogene. 2005;24:5053-68
67. Madar S, Brosh R, Buganim Y, Ezra O, Goldstein I, Solomon H. et al. Modulated expression of WFDC1 during carcinogenesis and cellular senescence. Carcinogenesis. 2009;30:20-7
68. Bouchard D, Morisset D, Bourbonnais Y, Tremblay GM. Proteins with whey-acidic-protein motifs and cancer. The Lancet Oncology. 2006;7:167-74
69. Li H, Fan X, Houghton J. Tumor microenvironment: the role of the tumor stroma in cancer. Journal of cellular biochemistry. 2007;101:805-15
70. Toole BP. Hyaluronan: from extracellular glue to pericellular cue. Nat Rev Cancer. 2004;4:528-39
71. Itano N, Zhuo L, Kimata K. Impact of the hyaluronan-rich tumor microenvironment on cancer initiation and progression. Cancer science. 2008;99:1720-5
72. Karjalainen JM, Tammi RH, Tammi MI, Eskelinen MJ, Agren UM, Parkkinen JJ. et al. Reduced level of CD44 and hyaluronan associated with unfavorable prognosis in clinical stage I cutaneous melanoma. The American journal of pathology. 2000;157:957-65
73. Misra S, Ghatak S, Toole BP. Regulation of MDR1 expression and drug resistance by a positive feedback loop involving hyaluronan, phosphoinositide 3-kinase, and ErbB2. The Journal of biological chemistry. 2005;280:20310-5
74. Misra S, Ghatak S, Zoltan-Jones A, Toole BP. Regulation of multidrug resistance in cancer cells by hyaluronan. The Journal of biological chemistry. 2003;278:25285-8
75. Stern R. Hyaluronan metabolism: a major paradox in cancer biology. Pathologie-biologie. 2005;53:372-82
76. Peng QL, Zhao LY, Hou YX, Sun Y, Wang LY, Luo HJ. et al. Biological Characteristics and Genetic Heterogeneity between Carcinoma-Associated Fibroblasts and Their Paired Normal Fibroblasts in Human Breast Cancer. Plos One. 2013:8
77. Nagasaki T, Hara M, Nakanishi H, Takahashi H, Sato M, Takeyama H. Interleukin-6 released by colon cancer-associated fibroblasts is critical for tumour angiogenesis: anti-interleukin-6 receptor antibody suppressed angiogenesis and inhibited tumour-stroma interaction. British journal of cancer. 2014;110:469-78
78. Paland N, Kamer I, Kogan-Sakin I, Madar S, Goldfinger N, Rotter V. Differential influence of normal and cancer-associated fibroblasts on the growth of human epithelial cells in an in vitro cocultivation model of prostate cancer. Molecular cancer research: MCR. 2009;7:1212-23
79. Mercier I, Casimiro MC, Wang CG, Rosenberg AL, Quong J, Minkeu A. et al. Human breast cancer-associated fibroblasts (CAFs) show caveolin-1 downregulation and RB tumor suppressor functional inactivation - Implications for the response to hormonal therapy. Cancer Biology & Therapy. 2008;7:1212-25
80. Flaberg E, Guven H, Savchenko A, Pavlova T, Kashuba V, Szekely L. et al. The architecture of fibroblast monolayers of different origin differentially influences tumor cell growth. International Journal of Cancer. 2012;131:2274-83
81. Albrengues J, Meneguzzi G, Gaggioli C. [Carcinoma-associated fibroblasts in cancer: the great escape]. Med Sci (Paris). 2014;30:391-7
82. Flach EH, Rebecca VW, Herlyn M, Smalley KS, Anderson AR. Fibroblasts contribute to melanoma tumor growth and drug resistance. Molecular pharmaceutics. 2011;8:2039-49
83. Godden JL, Edward M, MacKie RM. Melanoma cell-derived factor stimulation of fibroblast glycosaminoglycan synthesis--the role of platelet-derived growth factor. Eur J Cancer. 1999;35:473-80
84. Busse A, Keilholz U. Role of TGF-beta in melanoma. Current pharmaceutical biotechnology. 2011;12:2165-75
85. Choe C, Shin YS, Kim SH, Jeon MJ, Choi SJ, Lee J. et al. Tumor-stromal interactions with direct cell contacts enhance motility of non-small cell lung cancer cells through the hedgehog signaling pathway. Anticancer research. 2013;33:3715-23
86. Hsu MY, Meier FE, Nesbit M, Hsu JY, Van Belle P, Elder DE. et al. E-cadherin expression in melanoma cells restores keratinocyte-mediated growth control and down-regulates expression of invasion-related adhesion receptors. The American journal of pathology. 2000;156:1515-25
87. Tang A, Eller MS, Hara M, Yaar M, Hirohashi S, Gilchrest BA. E-Cadherin Is the Major Mediator of Human Melanocyte Adhesion to Keratinocytes in-Vitro. J Invest Dermatol. 1994;102:536
88. Li G, Satyamoorthy K, Herlyn M. N-cadherin-mediated intercellular interactions promote survival and migration of melanoma cells. Cancer Res. 2001;61:3819-25
89. Lade-Keller J, Riber-Hansen R, Guldberg P, Schmidt H, Hamilton-Dutoit SJ, Steiniche T. E- to N-cadherin switch in melanoma is associated with decreased expression of phosphatase and tensin homolog and cancer progression. Brit J Dermatol. 2013;169:618-28
90. Hao L, Ha JR, Kuzel P, Garcia E, Persad S. Cadherin switch from E- to N-cadherin in melanoma progression is regulated by the PI3K/PTEN pathway through Twist and Snail. Brit J Dermatol. 2012;166:1184-97
91. Kinugasa Y, Matsui T, Takakura N. CD44 expressed on cancer-associated fibroblasts is a functional molecule supporting the stemness and drug resistance of malignant cancer cells in the tumor microenvironment. Stem Cells. 2014;32:145-56
92. Spaeth EL, Labaff AM, Toole BP, Klopp A, Andreeff M, Marini FC. Mesenchymal CD44 Expression Contributes to the Acquisition of an Activated Fibroblast Phenotype via TWIST Activation in the Tumor Microenvironment. Cancer Res. 2013;73:5347-59
93. Shim S, Hah J, Sung M. Cancer-associated fibroblasts (CAF) expressing CD44 high increased the aggressiveness of cancer cells. AACR International Conference on Translational Cancer Medicine. San Francisco, CA: Clin Cancer Res. 2010;2010:A6
94. Lu P, Weaver VM, Werb Z. The extracellular matrix: a dynamic niche in cancer progression. The Journal of cell biology. 2012;196:395-406
95. Wandel E, Grasshoff A, Mittag M, Haustein UF, Saalbach A. Fibroblasts surrounding melanoma express elevated levels of matrix metalloproteinase-1 (MMP-1) and intercellular adhesion molecule-1 (ICAM-1) in vitro. Experimental dermatology. 2000;9:34-41
96. Ntayi C, Hornebeck W, Bernard P. Influence of cultured dermal fibroblasts on human melanoma cell proliferation, matrix metalloproteinase-2 (MMP-2) expression and invasion in vitro. Archives of dermatological research. 2003;295:236-41
97. Loffek S, Zigrino P, Angel P, Anwald B, Krieg T, Mauch C. High invasive melanoma cells induce matrix metalloproteinase-1 synthesis in fibroblasts by interleukin-1alpha and basic fibroblast growth factor-mediated mechanisms. J Invest Dermatol. 2005;124:638-43
98. Ma J, Tang X, Wong P, Jacobs B, Borden EC, Bedogni B. Noncanonical activation of Notch1 protein by membrane type 1 matrix metalloproteinase (MT1-MMP) controls melanoma cell proliferation. The Journal of biological chemistry. 2014;289:8442-9
99. Meierjohann S, Hufnagel A, Wende E, Kleinschmidt MA, Wolf K, Friedl P. et al. MMP13 mediates cell cycle progression in melanocytes and melanoma cells: in vitro studies of migration and proliferation. Molecular cancer. 2010;9:201
100. Huber MA, Kraut N, Park JE, Schubert RD, Rettig WJ, Peter RU. et al. Fibroblast Activation Protein: Differential Expression and Serine Protease Activity in Reactive Stromal Fibroblasts of Melanocytic Skin Tumors. J Investig Dermatol. 2003;120:182-8
101. Liu R, Li H, Liu L, Yu J, Ren X. Fibroblast activation protein: A potential therapeutic target in cancer. Cancer Biol Ther. 2012;13:123-9
102. Waster P, Rosdahl I, Gilmore BF, Seifert O. Ultraviolet exposure of melanoma cells induces fibroblast activation protein-alpha in fibroblasts: Implications for melanoma invasion. International journal of oncology. 2011;39:193-202
103. Ruby C, Kaufman H. Modulating the Tumor Microenvironment. In: (ed.) Gajewski TF, Hodi FS. Targeted Therapeutics in Melanoma. Springer New York. 2012:353-69
104. Comito G, Giannoni E, Di Gennaro P, Segura CP, Gerlini G, Chiarugi P. Stromal fibroblasts synergize with hypoxic oxidative stress to enhance melanoma aggressiveness. Cancer letters. 2012;324:31-41
105. Taddei ML, Giannoni E, Raugei G, Scacco S, Sardanelli AM, Papa S. et al. Mitochondrial Oxidative Stress due to Complex I Dysfunction Promotes Fibroblast Activation and Melanoma Cell Invasiveness. Journal of Signal Transduction. 2012;2012:10
106. Richmond A, Yang J, Su Y. The good and the bad of chemokines/chemokine receptors in melanoma. Pigment cell & melanoma research. 2009;22:175-86
107. Mishra P, Banerjee D, Ben-Baruch A. Chemokines at the crossroads of tumor-fibroblast interactions that promote malignancy. J Leukocyte Biol. 2011;89:31-9
108. Augsten M, Hagglof C, Olsson E, Stolz C, Tsagozis P, Levchenko T. et al. CXCL14 is an autocrine growth factor for fibroblasts and acts as a multi-modal stimulator of prostate tumor growth. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:3414-9
109. Murakami T, Maki W, Cardones AR, Fang H, Tun Kyi A, Nestle FO. et al. Expression of CXC chemokine receptor-4 enhances the pulmonary metastatic potential of murine B16 melanoma cells. Cancer Res. 2002;62:7328-34
110. Scala S, Ottaiano A, Ascierto PA, Cavalli M, Simeone E, Giuliano P. et al. Expression of CXCR4 predicts poor prognosis in patients with malignant melanoma. Clinical cancer research: an official journal of the American Association for Cancer Research. 2005;11:1835-41
111. Giuliano P Jr, Ierano C, Ascierto PA, Napolitano M, Lombardi M, Liguori M. et al. Melanoma associated fibroblasts promote melanoma invasiveness: Possible cross talk between CXCR4 and VEGF receptor. AACR Meeting Abstracts. 2006;2006:42-b
112. Cardones AR, Murakami T, Hwang ST. CXCR4 enhances adhesion of B16 tumor cells to endothelial cells in vitro and in vivo via beta(1) integrin. Cancer Res. 2003;63:6751-7
113. O'Boyle G, Swidenbank I, Marshall H, Barker CE, Armstrong J, White SA. et al. Inhibition of CXCR4-CXCL12 chemotaxis in melanoma by AMD11070. British journal of cancer. 2013;108:1634-40
114. Takekoshi T, Ziarek JJ, Volkman BF, Hwang ST. A locked, dimeric CXCL12 variant effectively inhibits pulmonary metastasis of CXCR4-expressing melanoma cells due to enhanced serum stability. Molecular cancer therapeutics. 2012;11:2516-25
115. Gallagher PG, Bao Y, Prorock A, Zigrino P, Nischt R, Politi V. et al. Gene Expression Profiling Reveals Cross-talk between Melanoma and Fibroblasts: Implications for Host-Tumor Interactions in Metastasis. Cancer Res. 2005;65:4134-46
116. Li L, Dragulev B, Zigrino P, Mauch C, Fox JW. The invasive potential of human melanoma cell lines correlates with their ability to alter fibroblast gene expression in vitro and the stromal microenvironment in vivo. International journal of cancer Journal international du cancer. 2009;125:1796-804
117. Terai M, Eto M, Young GD, Berd D, Mastrangelo MJ, Tamura Y. et al. Interleukin 6 mediates production of interleukin 10 in metastatic melanoma. Cancer immunology, immunotherapy: CII. 2012;61:145-55
118. Na YR, Lee JS, Lee SJ, Seok SH. Interleukin-6-induced Twist and N-cadherin enhance melanoma cell metastasis. Melanoma research. 2013;23:434-43
119. Casey TM, Eneman J, Crocker A, White J, Tessitore J, Stanley M. et al. Cancer associated fibroblasts stimulated by transforming growth factor beta1 (TGF-beta 1) increase invasion rate of tumor cells: a population study. Breast cancer research and treatment. 2008;110:39-49
120. Villanueva J, Herlyn M. Melanoma and the tumor microenvironment. Current oncology reports. 2008;10:439-46
121. Halaban R, Rubin JS, White W. met and HGF-SF in normal melanocytes and melanoma cells. Exs. 1993;65:329-39
122. Gaggioli C, Deckert M, Robert G, Abbe P, Batoz M, Ehrengruber MU. et al. HGF induces fibronectin matrix synthesis in melanoma cells through MAP kinase-dependent signaling pathway and induction of Egr-1. Oncogene. 2005;24:1423-33
123. Koefinger P, Wels C, Joshi S, Damm S, Steinbauer E, Beham-Schmid C. et al. The cadherin switch in melanoma instigated by HGF is mediated through epithelial-mesenchymal transition regulators. Pigment cell & melanoma research. 2011;24:382-5
124. Poser I, Dominguez D, de Herreros AG, Varnai A, Buettner R, Bosserhoff AK. Loss of E-cadherin expression in melanoma cells involves up-regulation of the transcriptional repressor Snail. The Journal of biological chemistry. 2001;276:24661-6
125. Cianfarani F, Mastroeni S, Odorisio T, Passarelli F, Cattani C, Mannooranparampil TJ. et al. Expression of vascular endothelial growth factor-C in primary cutaneous melanoma predicts sentinel lymph node positivity. Journal of cutaneous pathology. 2012;39:826-34
126. Francia G, Emmenegger U, Kerbel RS. Tumor-Associated Fibroblasts as "Trojan Horse" Mediators of Resistance to Anti-VEGF Therapy. Cancer Cell. 2009;15:3-5
127. Haass NK, Smalley KS, Herlyn M. The microenvironment is the key determinant of melanoma drug resistance. J Invest Dermatol. 2006;126:145
128. Castells M, Thibault B, Delord JP, Couderc B. Implication of Tumor Microenvironment in Chemoresistance: Tumor-Associated Stromal Cells Protect Tumor Cells from Cell Death. Int J Mol Sci. 2012;13:9545-71
129. Tredan O, Galmarini CM, Patel K, Tannock IF. Drug resistance and the solid tumor microenvironment. Journal of the National Cancer Institute. 2007;99:1441-54
130. Liao D, Reisfeld R. Targeting Tumor Associated Fibroblasts Tumor Associated Fibroblasts and Chemotherapy. In: (ed.) Mueller MM, Fusenig NE. Tumor-Associated Fibroblasts and their Matrix. Springer Netherlands. 2011:403-18
131. Loeffler M, Kruger JA, Niethammer AG, Reisfeld RA. Targeting tumor-associated fibroblasts improves cancer chemotherapy by increasing intratumoral drug uptake. Journal of Clinical Investigation. 2006;116:1955-62
132. Zigrino P, Nischt R, Mauch C. The disintegrin-like and cysteine-rich domains of ADAM-9 mediate interactions between melanoma cells and fibroblasts. The Journal of biological chemistry. 2011;286:6801-7
133. Tiago M, de Oliveira EM, Brohem CA, Pennacchi PC, Paes RD, Haga RB. et al. Fibroblasts Protect Melanoma Cells from the Cytotoxic Effects of Doxorubicin. Tissue engineering Part A. 2014;20:2412-21
134. Gonda TA, Varro A, Wang TC, Tycko B. Molecular biology of cancer-associated fibroblasts: Can these cells be targeted in anti-cancer therapy? Seminars in cell & developmental biology. 2010;21:2-10
135. Xing F, Saidou J, Watabe K. Cancer associated fibroblasts (CAFs) in tumor microenvironment. Front Biosci (Landmark Ed). 2010;15:166-79
136. Togo S, Polanska UM, Horimoto Y, Orimo A. Carcinoma-associated fibroblasts are a promising therapeutic target. Cancers. 2013;5:149-69
137. Liu ZJ, Li Y, Tan YR, Xiao M, Zhang JL, Radtke F. et al. Inhibition of Fibroblast Growth by Notch1 Signaling Is Mediated by Induction of Wnt11-Dependent WISP-1. Plos One. 2012:7
138. Lee J, Fassnacht M, Nair S, Boczkowski D, Gilboa E. Tumor immunotherapy targeting fibroblast activation protein, a product expressed in tumor-associated fibroblasts. Cancer Res. 2005;65:11156-63
139. Pietras K, Pahler J, Bergers G, Hanahan D. Functions of paracrine PDGF signaling in the proangiogenic tumor stroma revealed by pharmacological targeting. Plos Med. 2008;5:123-38
140. Bertolotto C. Melanoma: from melanocyte to genetic alterations and clinical options. Scientifica. 2013;2013:635203
Author contact
![]() Corresponding author: Yuhang Zhang, Ph.D., Division of Pharmaceutical Sciences, College of Pharmacy, University of Cincinnati, Cincinnati, OH 45267, USA. Tel: 513-558-0740; Fax: 513-558-4372; Email: yuhang.zhangedu
Corresponding author: Yuhang Zhang, Ph.D., Division of Pharmaceutical Sciences, College of Pharmacy, University of Cincinnati, Cincinnati, OH 45267, USA. Tel: 513-558-0740; Fax: 513-558-4372; Email: yuhang.zhangedu