Journal of Cancer
ISSN: 1837-9664
3.2
Impact Factor
ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2015; 6(7):623-631. doi:10.7150/jca.11291 This issue Cite
Research Paper
Platycodin-D Induced Autophagy in Non-Small Cell Lung Cancer Cells via PI3K/Akt/mTOR and MAPK Signaling Pathways
Ruolin Zhao1, 2*, Meijuan Chen2*, Zequn Jiang2, Fengming Zhao2, Beili Xi1, Xu Zhang2 ![]() , Haian Fu3, 4
, Haian Fu3, 4 ![]() , Kunfu Zhou1, 2
, Kunfu Zhou1, 2 ![]()
1. The Pre-clinical Medicine College, Nanjing University of Chinese Medicine, Nanjing 210023, China
2. Jiangsu Collaborative Innovation Center of Traditional Chinese Medicine (TCM) Prevention and Treatment of Tumor, Nanjing University of Chinese Medicine, Nanjing 210023, China
3. Department of Pharmacology, Emory University School of Medicine, Atlanta, GA 30322, USA
4. Department of Hematology & Medical Oncology, Emory University School of Medicine, Atlanta, GA 30322, USA
* Ruolin Zhao and Meijuan Chen contributed equally to this article.
Received 2014-12-9; Accepted 2015-4-2; Published 2015-5-23
Citation:
Zhao R, Chen M, Jiang Z, Zhao F, Xi B, Zhang X, Fu H, Zhou K. Platycodin-D Induced Autophagy in Non-Small Cell Lung Cancer Cells via PI3K/Akt/mTOR and MAPK Signaling Pathways. J Cancer 2015; 6(7):623-631. doi:10.7150/jca.11291. https://www.jcancer.org/v06p0623.htm
Other stylesAbstract
Platycodin-D (PD) is an effective triterpene saponin extracted from the root of Platycodon grandiflorum which has been used clinically to treat pulmonary diseases in traditional Chinese medicine. Recently, it has been reported that PD has anti-tumor effects in various cancer models through the induction of apoptosis. However, whether PD induces autophagy in both cell lines and its molecular mechanisms have not been elucidated. Here, our present study confirmed that PD induced autophagy in both NCI-H460 and A549 cells via up-regulating the expression levels of Atg-3, Atg-7 and Beclin-1. Meanwhile, PD contributed to the up-regulation of LC3-II at both protein and mRNA levels. Further detection of the PI3K/Akt/mTOR signaling pathway compared to LY294002 (PI3K kinase inhibitor), RAP (mTOR kinase inhibitor) and insulin (an activator of PI3K/Akt/mTOR signaling pathway) showed that PD induced autophagy through inhibiting the pathway at p-Akt (Ser473), p-p70S6K (Thr389) and p-4EBP1 (Thr37/46) in both cell lines. Moreover, the examination of MAPK signaling pathway showed that PD treatment increased the phosphorylation of JNK and p38 MAPK, while decreased the phosphorylation of Erk1/2 in both cell lines. Additionally, the effects assessed with a panel of pharmacologic inhibitors, including U0126 (Erk1/2 kinase inhibitor), SP600125 (JNK kinase inhibitor) and SB203580 (p38 MAPK kinase inhibitor) suggested that the activation of JNK and p38 MAPK participated in PD-induced autophagy. Taken together, these findings suggested that PD induced autophagy in NCI-H460 and A549 cells through inhibiting PI3K/Akt/mTOR signaling pathway and activating JNK and p38 MAPK signaling pathways. Therefore, PD may be an alternative compound for NSCLC therapy.
Keywords: Platycodin-D, non-small cell lung cancer, autophagy, PI3K/Akt/mTOR, MAPK signaling pathways
Introduction
PD is a triterpenoid saponin isolated from the root of Platycodon grandiflorum, which has been widely used in traditional Chinese medicine for treatment against various pulmonary diseases and respiratory disorders [1-3]. Recently, it has been extensively investigated as a potential alternative therapy for treatment of various cancers, including breast cancer, gastric cancer, leukemia, colon cancer and so on [1-3]. Our preliminary studies have demonstrated that PD has an anti-NSCLC effect associated with the induction of apoptosis, however, whether PD induces autophagy in NSCLC cells has yet to be identified.
Most anticancer drugs are associated with the induction of programmed cell death (PCD). Accumulating studies have suggested that autophagy (Type II PCD), consistent with apoptosis (Type I PCD), is also important for the regulation of cancer development and progression, and may be regarded as a potential therapeutic approach for anticancer researches [4]. However, recent evidence has indicated that apoptosis-induced agents may confer drug-resistance following the anticancer effects especially at the late stage of tumor development. Thus, autophagy which is an evolutionarily conserved catabolic process of self-degradation of organelles and cytosolic macromolecules has been considered as an alternative therapeutic approach in cancer cells [5]. Autophagy initiation which completed with the accumulation of the ULK1/2-ATG13-FIP200 complex results in development of the isolation membrane, also known as a phagosome. On receiving autophagic stimulus, the phagosome converts to autophagosome depending on autophagy-related genes (Atg) proteins, then its maturation is completed upon fusion with lysosome to form autophagolysosome [6].
Several signaling pathways have been demonstrated to interfere with autophagy. Among them, phosphatidylinositol 3-kinase/protein kinase B/mammalian target of rapamycin (PI3K/Akt/mTOR) and mitogen-activated protein kinase (MAPK) signaling pathways especially play a critical role in regulating establishment of autophagy in cancer cells [7-9]. Recent studies have demonstrated the constitutive activation of the PI3K/Akt/mTOR signaling pathway occurs in 90% of NSCLC cell lines, and the inhibition of this signaling cascade is not only important for induction of autophagic cell death but also for developing new treatments for NSCLC [10]. Consistent with the PI3K/Akt/mTOR signaling pathway, MAPK signaling pathways including extracellular signal-regulated kinase (Erk), p38 MAPK and c-jun NH2-terminal kinase (JNK) have been identified as chemotherapeutic targets for sensitizing cancer cells to autophagy [7, 8].
In this study, we aimed to investigate the effects of PD-induced autophagy in NCI-H460 and A549 cell lines and its underlying mechanisms focusing on the role of PI3K/Akt/mTOR and MAPK signaling pathways in PD-induced autophagy.
Materials and methods
Reagents
Platycodin-D (PD) (MW: 1224.58, HPLC ≥ 98%) was purchased from Shanghai Yuanye biotechnology Co. Ltd. (Shanghai, China). The chemicals used were rapamycin, LY294002, U0126, SP600125 and SB203580 (Cell Signaling Technology, USA), insulin, Gimesa (Sigma, USA), TRIzol reagent (Invitrogen, USA), Real-time PCR Master Mix (Toyobo, Japan). Mouse- or rabbit-polyclonal antibodies specific for LC3-І /II, Beclin-1, Atg-5 and Atg-7, p-Akt (Ser473), p-p70S6K (Thr389), p-4EBP1 (Thr37/46), Akt, p70S6K, 4EBP1, p-Erk1/2, p-JNK, p-p38 MAPK, β-actin and secondary antibodies (goat anti-mouse or anti-rabbit IgG-conjugated horseradish peroxidase (HRP)) were all purchased from Cell Signaling Technology (Beverly, USA).
Cell culture
Human NSCLC cells lines (A549 and NCI-H460) were obtained from the Institute of Biochemistry and Cell Biology (Shanghai, China). NCI-H460 and A549 cells were cultured in DMEM and F12 medium (HyClone, USA) respectively, supplemented with 10% fetal bovine serum (FBS) (Gibco, USA), 100 U/ml penicillin-streptomycin mixed antibiotics at 37°C in a humidified 5% CO2 incubator.
Transmission electron microscopy (TEM)
After being exposed to 20 or 30 µmol/L of PD for 24 h, the cells were trypsinized, washed with PBS and fixed in 2.5% glutaraldehyde in 0.1 mol/L phosphate buffer (pH 7.2) overnight at 4°C. The next day, cells were washed three times with 0.1 mol/L phosphate buffer. They were then fixed in 1% aqueous osmium, dehydrated with increasing concentrations of ethanol (30, 50, 70, 80, 90 and 100%), and embedded in araldite. Ultrathin sections were prepared with a microtome (Leica, Germany) and mounted on copper grids. The samples were stained with 2% aqueous uranyl acetate and lead citrate and observed in a transmission electron microscope (TEM; Jeol, Japan).
Western blot analysis
For western blot analysis, cells were lysed in RIPA buffer containing 50 mmol/L Tris/HCl (pH 8.0), 150 mmol/L NaCl, 1% Nonidet-P40, 1% sodium deoxycholate, 0.1% SDS, 0.1 mmol/L DTT, 0.05 mmol/L PMSF, 0.002 mg/ml aprotinin, 0.002 mg/ml leupeptin, and 1 mmol/L NaVO3. The protein concentration of each supernatant was determined by the BCA protein assay. Equal amounts of protein were loaded and separated by 10 or 12% SDS-PAGE and then transferred onto polyvinylidene difluoride membranes. The membranes were incubated overnight with appropriate primary antibodies against LC3-І /II, Beclin-1, Atg-5 and Atg-7, p-Akt (Ser473), p-p70S6K (Thr389), p-4EBP1 (Thr37/46), Akt, p70S6K, 4EBP1, p-Erk1/2 , p-JNK, p-p38 MAPK or β-actin overnight at 4˚C, and then with HRP-conjugated secondary antibodies (anti-rabbit or mouse immunoglobulin G) for an additional 1 h at room temperature. Immunoreactivity was detected by enhanced chemiluminescence (ECL) (Bio-Rad, USA). β-actin was used as a loading control. Quantitative analysis was performed by Image Lab™ software.
Quantitative reverse transcription-PCR for mRNA quantification
Total RNA was extracted using the TRIzol reagent (Invitrogen, USA), and cDNA was prepared using 0.5μg of oligo (dT) primers and the PrimeScript RT reagent (Toyobo, Japan) according to the manufacturer's protocol. Quantitative reverse transcription-PCR (QRT-PCR) analysis was performed using SYBR green assays (TaKaRa) on the following genes using the following primers: LC3-II primer, Sense primer: 5'-AAACGCATTTGCCATCACAGT-3' and Antisense primer: 5'-GTGAGGACTTTGGGTGTGGTTC-3'; Human-GAPDH primer, Sense primer: 5'- TGGTATCGTGGAAGGACTCA-3', Antisense primer: 5'- CCAGTAGAGGCAGGGATGAT-3'. Each test was carried out in triplicate according to standard protocol. Data were calculated using the 2ΔΔCt method comparing ΔCt of treated A549 cells to ΔCt of control untreated samples. Reactions were incubated in the LightCycler 480 QRT-PCR system. Ct values were calculated using the SDS software version 2.3 applying automatic baseline and threshold settings.
Statistical analysis
All quantitative data presented were the mean ± SD from at least three independent experiments. The SPSS 19.0 software package was used to perform all statistical analysis. Comparisons between two groups were performed using the Student's-t test and between multiple groups using ANOVA analysis. A value of P < 0.05 was considered statistically significant.
Results
Morphological changes of autophagy-induced by PD in NCI-H460 and A549 cells
NCI-H460 and A549 cells were treated with 0, 5, 10, 20 or 30 μmol/L of PD, respectively. After 24 h treatment, cells stained with Gimesa were observed using phase contrast microscopy. With the increasing concentrations of PD, cells had shrunk, accumulated vacuoles in the cytoplasm, and cell density significantly decreased compared with untreated control group (Fig. 1 A).
Figure 1
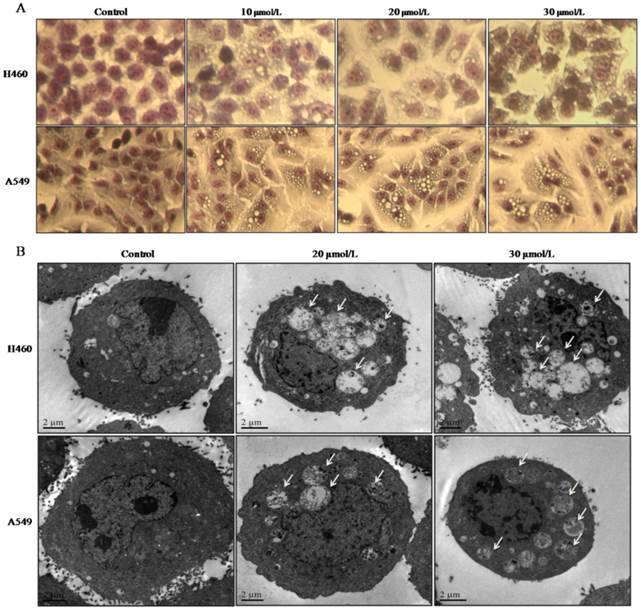
PD induced morphological changes of NCI-H460 and A549 cells. (A) NCI-H460 and A549 cells treated with PD at various concentrations of 0, 10, 20, and 30 μmol/L, respectively. After 24 h treatment, cells stained with Gimesa were observed using phase-contrast microscopy (×400). (B) NCI-H460 and A549 cells were exposed to 0, 20 and 30 μmol/L of PD for 24h followed by observation using a transmission electron microscope (TEM). Numerous autophagosomes with typical double-layer membranes containing organelle remnants were highlighted by arrows.
Transmission electron microscopy (TEM) is a conventional method for monitoring autophagy. Through TEM detection, we found cytoplasmic vaculoes in both NCI- H460 and A549 cells after exposure to 20 or 30 μmol/L of PD for 24 h, and the cytoplasmic vaculoes had double-layered membranes and many of them contained cytoplasmic organelles or myelin figures (Fig. 1 B). Particularly, with the increasing concentrations of PD treatment, the vacuoles increased in size and number and fused into larger vacuoles compared with the untreated control group (Fig. 1 B). These morphological changes indicated that PD induced autophagosome formation. Thus, we speculated the treatment with PD might induce autophagy in both cell lines.
PD induced autophagy in NCI-H460 and A549 cells
To confirm the exact effects of PD on induction of autophagy in NCI- H460 and A549 cells, autophagy-related genes proteins, which are called Atg proteins including LC3-I/II (Atg-8), Beclin-1 (Atg-6), Atg-3 and Atg-7 were detected by western blot analysis. Our data showed that with PD treatment, the expression of Beclin-1, Atg-3 and Atg-7 and the conversion of LC3-I to LC3-II increased in a dose- and time-dependent manner (Fig. 2 A-D). We next examined the expression of LC3-II, which serves as an ultimate biomarker of autophagy, at the mRNA level by using qRT-PCR. The data in Fig. 2 E demonstrated that the mRNA level of LC3-II was dramatically up-regulated after 20 or 30 μmol/L of PD treatment for 24 h (P < 0.01, P < 0.05, respectively). Collectively, these results provided evidence that PD induced autophagy in NCI- H460 and A549 cell lines.
Figure 2
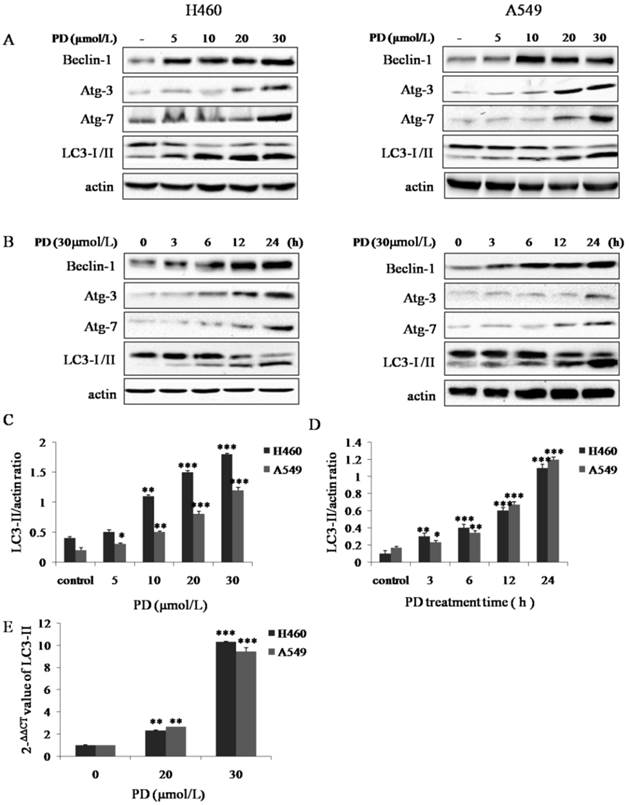
Effect of PD on inducing autophagy in NCI-H460 and A549 cells. (A and B) NCI-H460 and A549 cells treated with 0, 5, 10, 20 and 30 µmol/L of PD for 24 h or 30 µmol/L of PD for 0, 3, 6, 12 and 24 h were analyzed by western-blot with antibodies against LC3-I/II, Beclin-1, Atg-3 and Atg-7. (C and D) Densitometry analysis of LC3-II level relative to actin was performed. (E) The mRNA expression level of LC3-II induced by PD in both cell lines was detected by Quantitative reverse transcription-PCR analysis. Representative results of three independent experiments are shown. β-actin was used as a loading control. Error bars, SD; *, P<0.05; **, P<0.01, ***, P<0.001 versus control values.
Effects of PD on PI3K/Akt/mTOR signaling pathway for induction of autophagy in NCI-H460 and A549 cells
The PI3K/Akt/mTOR signaling pathway plays a critical role in cell proliferation and autophagy. To better understand the molecular mechanisms of PD-induced autophagy, we determined the possible involvement of this signaling pathway. As shown in Fig. 3 A and B, the expression levels of p-Akt (Ser473), p-p70S6K (Thr389), and p-4EBP1 (Thr37/46) in both NCI-H460 and A549 cells were significantly inhibited by PD in a dose- and time-dependent manner.
We further investigated the effect of PD on the PI3K/Akt/mTOR/p70S6K/4EBP1 pathway compared with LY294002 (an inhibitor of PI3K) and RAP (an inhibitor of mTOR)[11, 12]. Similarly to LY294002, PD inhibited p-Akt (Ser473), meanwhile, it weakened the feedback activation of RAP on p-Akt (Ser473) in both cell lines (Fig. 3 C and D). Notably, the ratio of LC3-II/actin was enhanced by co-treatment with PD and LY294002 or PD and rapamycin compared with PD, LY294002 or RAP treatment alone (P<0.05) (Fig. 3 C, D and G). Thus, PD had a synergistic effect on induction of autophagy with LY294002 or RAP.
Insulin not only activates PI3K/Akt/mTOR signaling pathway but also suppresses autophagy [13, 14]. As shown in Fig. 3 E and F, 30 min of insulin treatment significantly phosphorylated Akt (Ser473), p70S6K (Thr389), and 4EBP1 (Thr37/46). In contrast, when cells were pretreated with PD and then stimulated with insulin, the phosphorylation levels of them were significantly inhibited. Meanwhile, LC3-II/actin ratio in both cell lines was suppressed by insulin, however, this effect of insulin was reduced by PD pretreatment (P<0.01) (Fig.3 E, F and H).
Taken together, it was clearly suggested that PD induced autophagy in NCI-H460 and A549 cells by inhibiting the PI3K/Akt/mTOR signaling pathway.
Effects of PD on MAPK signaling pathways for induction of autophagy in NCI-H460 and A549 cells
It has been suggested that MAPK signaling pathways are important for regulation of proliferation, differentiation, apoptosis, autophagy and cell cycle arrest [9, 11]. To investigate the role of MAPK signaling pathways in PD-induced autophagy, the activation of JNK, Erk1/2 and p38 MAPK was examined by Western blot assay using phosphorylated antibodies specific for the active forms of these kinases. The expression levels of p-p38 MAPK and p-JNK were activated by PD in a dose and time-dependent manner, however, p-Erk1/2 was inhibited by PD dose and time-dependently (Fig. 4 A and B ).
To further clarify whether PD-induced autophagy depended on p-p38 MAPK and p-JNK activation and p-Erk1/2 suppression, cells were treated with U0126 (an inhibitor of Erk1/2), SP600125 (an inhibitor of JNK) or SB203580 (an inhibitor of p38) for 4 h followed by treatment with or without PD for 24h. Our date showed that the ratio of LC3-II/LC3-I was dramatically increased in PD co-treatment with SP600125 or SB203580 group compared with that in SP600125 or SB203580 treatment group (P<0.01) (Fig. 4 C-F), suggesting the activation of p-JNK or p-p38 MAPK was involved in cell autophagy. Though the ratio of LC3-II/LC3-I in U0126 treatment group was increased, the ratio of LC3-II/LC3-I in PD co-treatment with U0126 group was lower than that by PD treatment alone (Fig. 4 F), suggesting inhibition of p-Erk1/2 was not involved in cell autophagy.
Taken together, these results demonstrated that PD induced autophagy in NCI-H460 and A549 cells through activation of p-p38 MAPK and p-JNK, while inhibition of p-Erk1/2 was not responsible for PD-induced autophagy.
Discussion
Currently, it has been reported that PD effectively induces apoptosis in many cancer cells [1-3]. And our previous studies had found PD significantly inhibited cell proliferation and viability in NSCLC cell lines NCI-H460 and A549 in a dose- and time-dependent manner, and it induced apoptosis through mitochondrial-dependent and Erk signaling pathways (under submitting). However, whether PD induces autophagy and its molecular mechanisms remains unknown.
Autophagy is considered an alternative therapeutic approach in cancer cells [5, 15]. Autophagic processes start with the formation of autophagosome, which is enclosed within double-membrane that entraps cytoplasmic components. Then, autophagosome fuse with lysosome to generate autophaglysome, where cytoplasmic molecules and organelles are degraded by lysosomal hydrolyases (Fig. 5) [16]. Initially, we found that most of the cells exposed to PD for 24 h had shrunk, accumulated vacuoles in the cytoplasm, and cell density decreased compared with the untreated control in dose-dependent manner (Fig. 1 A). Cell morphology observed with TEM showed that he cytoplasmic vaculoes had double-layered membranes and only some cytoplasmic organelles or myelin figures remained in both cell lines after 24 h treatment of PD (Fig. 1 B).
Figure 3
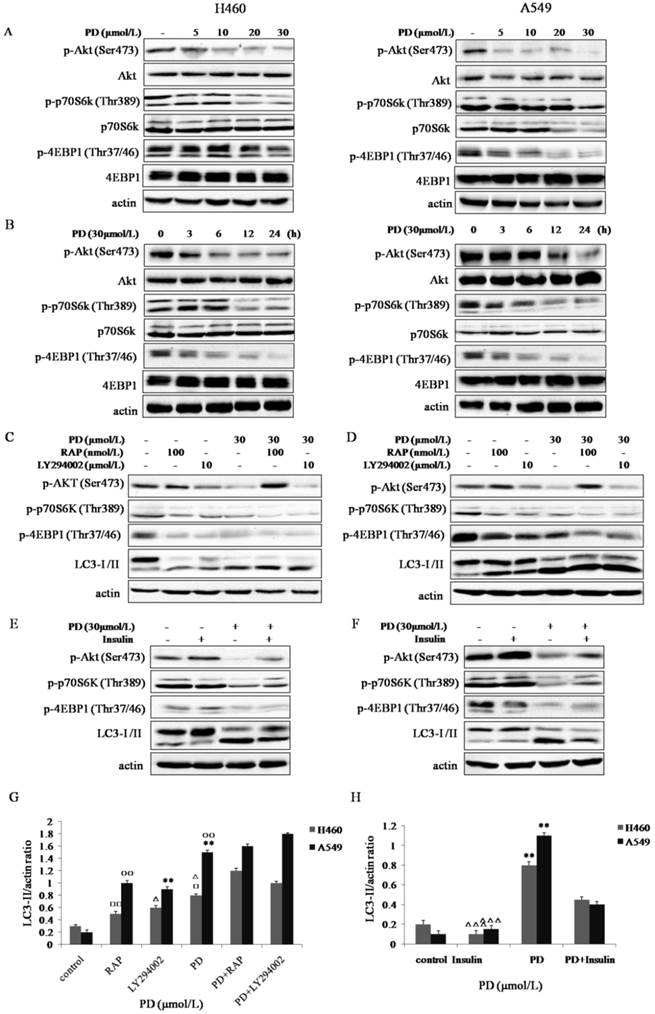
Effect of PD on PI3K/Akt/mTOR/p70s6k/4EBP1 signaling pathway in NCI-H460 and A549 cells. (A and B) NCI-H460 and A549 cells treated with 0, 5, 10, 20 and 30 µmol/L of PD for 24 h or 30 µmol/L of PD for 0, 3, 6, 12 and 24 h were analyzed by western-blot with antibodies against p-Akt (Ser473), p-p70S6K(Thr389), p-4EBP1 (Thr37/46), Akt, p70S6K and 4EBP1. (C and D) NCI-H460 and A549 cells treated with 30 µmol/L of PD, 10 µmol/L of LY294002 or 10 µmol/L of Rapmycin for 24 h were analyzed by western-blot with antibodies against p-Akt (Ser473), p-p70S6K (Thr389), p-4EBP1 (Thr37/46) and LC3-І /II. (E and F) Cells treated with 30 µmol/L of PD for 24 h followed by treatment with or without 200 nmol/L of insulin for 30 min were analyzed by western-blot for the expression levels of p-Akt (Ser473), p-p70S6K (Thr389) and LC3-І/II. (G and H) Densitometry analysis of LC3-II level relative to actin was performed in both two cell lines.○, □: P<0.05; and ○○, □□: P<0.01 versus PD + RAP values; *, △: P<0.05; and **, △△: P<0.01 versus PD + LY294002 values. △△△: P<0.001 Insulin versus PD + Insulin values; **: P<0.01 PD versus control values. Representative results of three independent experiments are shown. β-actin was used as a loading control. Error bars, SD.
Figure 4
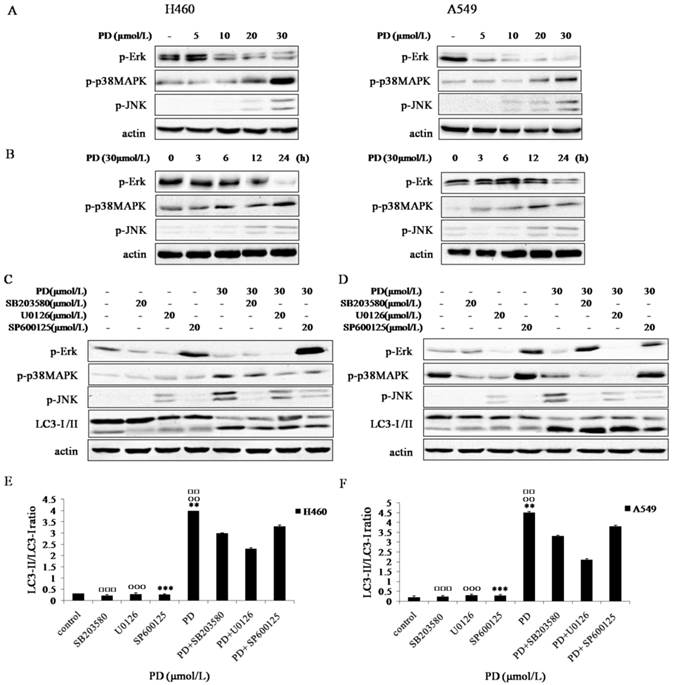
Effect of PD on MAPK signaling pathways in NCI-H460 and A549 cells. (A and B) NCI-H460 and A549 cells treated with 0, 5, 10, 20 and 30 µmol/L of PD for 24 h or 30 µmol/L of PD for 0, 3, 6, 12 and 24 h were analyzed by western blot with antibodies against p-Erk1/2, p-JNK and p-p38 MAPK. (C and D) NCI-H460 and A549 cells treated with 20 µmol/L of SB203580, 20 µmol/L of U0126 or 20 µmol/L of SP600125 for 4 h followed by treatment with or without 30 µmol/L of PD for 24 h were analyzed by western blot with antibodies against p-Erk 1/2 , p-JNK, p-p38 MAPK and LC3-І /II. (E and F) Densitometry analysis of LC3-II levels relative to LC3-І in NCI-H460 and A549 cells was performed. Representative results of three independent experiments are shown. β-actin was used as a loading control. Error bars, SD; □□: P<0.01 and □□□: P<0.001: versus PD + SB203580 values; ○○: P<0.01 and ○○○: P<0.001 versus PD + U0126 values; **: P<0.01 and ***: P<0.001 versus PD + SP600125 values.
Among the processes of autophagy, autophagosome-formation is regulated by Atg proteins which are essential to regulate chemotherapy-induced autophagy encoded by the autophagy-related genes [17]. There are two ubiqutin-like conjugation systems about Atg proteins. The first one results in covalent attachment of the ubiquitin-like protein Atg-12-Atg-5 (together with Atg-16) catalyzed by Atg-7 [18]. In another system, the LC3-I/ II (Atg-8) gets activated by Atg-7 and then undergoes lipidation with phosphatidylethanolamine (PE) by Atg-3 [19]. Moreover, Beclin-1 (Atg-6), a part of a class III PI3K complex, is necessary to form pre-autophagosomal structures [20]. Followed by the effects of Atg proteins, the cytoplasmic targets for autophagy are encapsulated in the autophagosome [4]. Our results indicated that PD induced autophagy as evidence by increasing the conversion of LC3-I to LC3-II and the accumulation of Atg-3, Atg-7 and Beclin-1 in a dose- and time-dependent manner (Fig. 2 A and B). LC3-II, recruited to forming autophagosomes, is an ultimate biomarker of autophagy [19]. Our data suggested that PD up-regulated the expression of LC3-II in both protein and mRNA levels in both cell lines (Fig. 2 C-E). Collectively, these results provided obvious evidence that PD induced autophagy in NCI-H460 and A549 cells.
Accumulating researches have highlighted that the inhibition of Akt and its downstream target p70S6K contribute to the initiation of autophagy, and the expression level of p-p70S6K could be utilized as a marker for mTOR activity. And the direct downstream target of p70S6K is 4EBP1 [21, 22]. As shown in Fig. 3 A and B, PD significantly decreased the expression levels of p-Akt (Ser473), p-p70S6K (Thr389) and p-4EBP1 (Thr37/46) in NCI-H460 and A549 cells. Compared with LY294002, which inhibits autophagy by the inhibition of class III PI3K [10], and RAP, which inhibits mTOR but has a negative feedback on Akt [23], PD decreased the activation of rapamycin on p-Akt (Ser473). Furthermore, the ratio of LC3-II/actin was enhanced by PD co-treatment with LY294002 or RAP compared with that by PD, LY294002 or RAP treatment alone (P<0.05), indicating that PD had a synergistic effect on induction of autophagy with LY294002 or RAP (Fig. 3 C, D and G). Subsequently, we used insulin to further clarify the correlation between insulin-mediated activation of the PI3K/Akt/mTOR pathway and PD-induced autophagy. Our results provided evidence that insulin activated this signaling pathway leading to autophagy suppression, the effect of insulin was abolished by PD addition (Fig.3 E and F). These results suggested that PD-induced autophagy in NCI-H460 and A549 cells was associated with the inhibition of PI3K/Akt/mTOR signaling pathway.
MAPK signaling pathways which consist of Erk, JNK and p38 MAPK signaling pathways are involved in many cellular processes including cell growth, proliferation, autophagy and apoptosis. The JNK, Erk1/2 and p38 MAPK, which are activated by phosphorylation, are key regulatory proteins in these pathways. We observed that the expression levels of p-p38 MAPK and p-JNK were activated by PD in a dose and time-dependent manner, while the expression level of p-Erk1/2 was inhibited by PD (Fig. 4 A and B ). To further clarify whether PD-induced autophagy was connected with p-p38 MAPK and p-JNK activation and p-Erk1/2 suppression, cells were pretreated with the pharmacological inhibitors SB203580, SP600125 or U0126 respectively, and then treated with or without PD. Notably, we found the conversion of LC3 І to LC3-II in PD co-treatment with SP600125 or SB203580 group was dramatically increased compared with that in SP600125 or SB203580 treatment group (P<0.01) (Fig. 4 C-F). Activation of the targeting JNK and p38 MAPK has been identified as chemotherapeutic agents for sensitizing cancer cells to autophagy [7, 24, 25]. In accordance with these findings, our results demonstrated the activation of JNK and p38 MAPK was involved in PD-induced autophagy in NCI-H460 and A549 cells. However, we also found the ratio of LC3-II/LC3-I in PD co-treatment with U0126 group was lower than that treated by PD alone, indicating that p-Erk1/2 was not responsible for PD-mediated autophagy (Fig. 4 F). And our previous study discovered that the inhibition of Erk signaling pathway by PD treatment induced apoptosis in NCI-H460 and A549 cells. Coordinately, increasing reports elucidate that among MAPK families Erk pathway is closely related with apoptosis [26, 27].
Figure 5
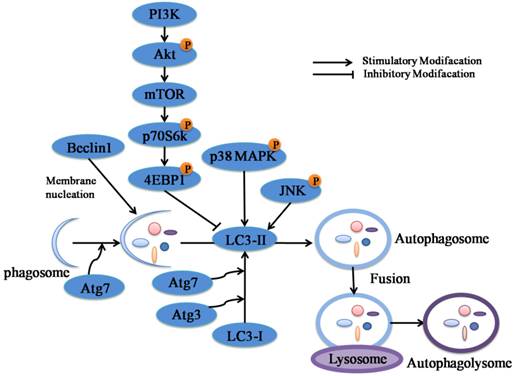
A schematic model of the molecular mechanisms associated with PD-induced autophagy in NCI-H460 and A549 cells. According to this model, when NCI-H460 and A549 cells were exposed to PD, the phagosome converted to double-layered membranes of autophagosomes through increasing expression levels of Atg proteins including Beclin-1, Atg-3 and Atg-7 as well as leading to the conversion of LC3-I to LC3-II. The inhibition of PI3K/Akt/mTOR signaling pathway and activation of JNK and p38 MAPK signaling pathways contributed to the accumulation of LC3-II, which were the possible upstream signaling pathways of PD-induced autophagy. Once the autophagosome was developed, its maturation was complete upon fusion with lysosome to form autophagolysosome. Eventually, programmed cell death was induced by PD. The schematic model compiled with the results and conclusions of this study (→: Stimulatory Modification, ⊥: Inhibitory Modification).
In conclusion, our experiments provided the first evidence that PD induced autophagy via inhibiting the PI3K/Akt/mTOR signaling pathway and activating JNK and p38 MAPK signaling pathways in NSCLC cells. Therefore, we speculated that PD may be an alternative therapeutic agent in the treatment of NSCLC. However, the intricate relationship between autophagy and apoptosis poses a big challenge for cancer treatment, as well as the cross-talk between PI3K/Akt/mTOR and MAPK signaling associated with the function of autophagy is uncertain. Hence, further studies are required to investigate above relationships by PD treatment and anti-NSCLC effects of PD in vivo.
Acknowledgements
This study was supported by a project funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD), grants from the People Programme (Marie Curie Actions) of the European Union's Seventh Framework Programme FP7/2007-2013/ under REA grant agreement n° PIR SES-GA-2013-612589", the U.S. National Institutes of Health grants P01 CA116676 and the Natural Science Foundation of Jiangsu Province (BK20131415).
Conflict of Interest
The authors have declared that no conflict of interest exists.
References
1. Chun J, Joo EJ, Kang M. et al. Platycodin D induces anoikis and caspase-mediated apoptosis via p38 MAPK in AGS human gastric cancer cells. Journal of cellular biochemistry. 2013;114(2):456-470
2. Yu JS, Kim AK. Platycodin D induces apoptosis in MCF-7 human breast cancer cells. Journal of medicinal food. 2010;13(2):298-305
3. Kim MO, Moon DO, Choi YH. et al. Platycodin D induces mitotic arrest in vitro, leading to endoreduplication, inhibition of proliferation and apoptosis in leukemia cells. International journal of cancer Journal international du cancer. 2008;122(12):2674-2681
4. Janku F, McConkey DJ, Hong DS. et al. Autophagy as a target for anticancer therapy. Nature reviews Clinical oncology. 2011;8(9):528-539
5. Chen S, Rehman SK, Zhang W. et al. Autophagy is a therapeutic target in anticancer drug resistance. Biochimica et biophysica acta. 2010;1806(2):220-229
6. Yang ZJ, Chee CE, Huang S. et al. The role of autophagy in cancer: therapeutic implications. Molecular cancer therapeutics. 2011;10(9):1533-1541
7. Sui X, Kong N, Ye L. et al. p38 and JNK MAPK pathways control the balance of apoptosis and autophagy in response to chemotherapeutic agents. Cancer letters. 2014;344(2):174-179
8. Sun Y, Zou M, Hu C. et al. Wogonoside induces autophagy in MDA-MB-231 cells by regulating MAPK-mTOR pathway. Food and chemical toxicology: an international journal published for the British Industrial Biological Research Association. 2013;51:53-60
9. Saiki S, Sasazawa Y, Imamichi Y. et al. Caffeine induces apoptosis by enhancement of autophagy via PI3K/Akt/mTOR/p70S6K inhibition. Autophagy. 2011;7(2):176-187
10. Chen M, Du Y, Qui M. et al. Ophiopogonin B-induced autophagy in non-small cell lung cancer cells via inhibition of the PI3K/Akt signaling pathway. Oncology reports. 2013;29(2):430-436
11. Vlahos CJ, Matter WF, Hui KY. et al. A specific inhibitor of phosphatidylinositol 3-kinase, 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002). The Journal of biological chemistry. 1994;269(7):5241-5248
12. Kunz J, Henriquez R, Schneider U. et al. Target of rapamycin in yeast, TOR2, is an essential phosphatidylinositol kinase homolog required for G1 progression. Cell. 1993;73(3):585-596
13. Yao H, Han X, Han X. The cardioprotection of the insulin-mediated PI3K/Akt/mTOR signaling pathway. American journal of cardiovascular drugs: drugs, devices, and other interventions. 2014;14(6):433-442
14. Garami A, Zwartkruis FJ, Nobukuni T. et al. Insulin activation of Rheb, a mediator of mTOR/S6K/4E-BP signaling, is inhibited by TSC1 and 2. Molecular cell. 2003;11(6):1457-1466
15. Beljanski V, Knaak C, Smith CD. A novel sphingosine kinase inhibitor induces autophagy in tumor cells. The Journal of pharmacology and experimental therapeutics. 2010;333(2):454-464
16. Mizushima N, Levine B, Cuervo AM. et al. Autophagy fights disease through cellular self-digestion. Nature. 2008;451(7182):1069-1075
17. Klionsky DJ, Abdalla FC, Abeliovich H. et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2012;8(4):445-544
18. Bommareddy A, Hahm ER, Xiao D. et al. Atg5 regulates phenethyl isothiocyanate-induced autophagic and apoptotic cell death in human prostate cancer cells. Cancer research. 2009;69(8):3704-3712
19. Mizushima N, Yoshimori T, Ohsumi Y. The role of Atg proteins in autophagosome formation. Annual review of cell and developmental biology. 2011;27:107-132
20. Kabeya Y, Mizushima N, Yamamoto A. et al. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. Journal of cell science. 2004;117(Pt 13):2805-2812
21. Kang R, Zeh HJ, Lotze MT. et al. The Beclin 1 network regulates autophagy and apoptosis. Cell death and differentiation. 2011;18(4):571-580
22. Viola G, Bortolozzi R, Hamel E. et al. MG-2477, a new tubulin inhibitor, induces autophagy through inhibition of the Akt/mTOR pathway and delayed apoptosis in A549 cells. Biochemical pharmacology. 2012;83(1):16-26
23. Rong Y, McPhee CK, Deng S. et al. Spinster is required for autophagic lysosome reformation and mTOR reactivation following starvation. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(19):7826-7831
24. O'Reilly KE, Rojo F, She QB. et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer research. 2006;66(3):1500-1508
25. Zhang C, Yang L, Wang XB. et al. Calyxin Y induces hydrogen peroxide-dependent autophagy and apoptosis via JNK activation in human non-small cell lung cancer NCI-H460 cells. Cancer letters. 2013;340(1):51-62
26. Webber JL. Regulation of autophagy by p38alpha MAPK. Autophagy. 2010;6(2):292-293
27. Cagnol S, Chambard JC. ERK and cell death: mechanisms of ERK-induced cell death--apoptosis, autophagy and senescence. The FEBS journal. 2010;277(1):2-21
28. Kohno M, Tanimura S, Ozaki K. Targeting the extracellular signal-regulated kinase pathway in cancer therapy. Biological & pharmaceutical bulletin. 2011;34(12):1781-1784
Author contact
![]() Corresponding authors: Dr Xu Zhang, Jiangsu Collaborative Innovation Center of Traditional Chinese Medicine (TCM) Prevention and Treatment of Tumor, Nanjing University of Chinese Medicine, Nanjing 210023, China. E-mail: zhangxutcmcom. Dr HaiAn Fu, Departments of Pharmacology and Hematology & Medical Oncology, Emory University School of Medicine, Atlanta, GA 30322, USA. E-mail: hfuedu. Dr Kunfu Zhou, The Pre-clinical Medicine College, Nanjing University of Chinese Medicine, Nanjing 210023, China. Tel: + 86-025-85811037, Fax: + 86-025-85811040. E-mail:zhoukunfucom
Corresponding authors: Dr Xu Zhang, Jiangsu Collaborative Innovation Center of Traditional Chinese Medicine (TCM) Prevention and Treatment of Tumor, Nanjing University of Chinese Medicine, Nanjing 210023, China. E-mail: zhangxutcmcom. Dr HaiAn Fu, Departments of Pharmacology and Hematology & Medical Oncology, Emory University School of Medicine, Atlanta, GA 30322, USA. E-mail: hfuedu. Dr Kunfu Zhou, The Pre-clinical Medicine College, Nanjing University of Chinese Medicine, Nanjing 210023, China. Tel: + 86-025-85811037, Fax: + 86-025-85811040. E-mail:zhoukunfucom
Citation styles
APA
Zhao, R., Chen, M., Jiang, Z., Zhao, F., Xi, B., Zhang, X., Fu, H., Zhou, K. (2015). Platycodin-D Induced Autophagy in Non-Small Cell Lung Cancer Cells via PI3K/Akt/mTOR and MAPK Signaling Pathways. Journal of Cancer, 6(7), 623-631. https://doi.org/10.7150/jca.11291.
ACS
Zhao, R.; Chen, M.; Jiang, Z.; Zhao, F.; Xi, B.; Zhang, X.; Fu, H.; Zhou, K. Platycodin-D Induced Autophagy in Non-Small Cell Lung Cancer Cells via PI3K/Akt/mTOR and MAPK Signaling Pathways. J. Cancer 2015, 6 (7), 623-631. DOI: 10.7150/jca.11291.
NLM
Zhao R, Chen M, Jiang Z, Zhao F, Xi B, Zhang X, Fu H, Zhou K. Platycodin-D Induced Autophagy in Non-Small Cell Lung Cancer Cells via PI3K/Akt/mTOR and MAPK Signaling Pathways. J Cancer 2015; 6(7):623-631. doi:10.7150/jca.11291. https://www.jcancer.org/v06p0623.htm
CSE
Zhao R, Chen M, Jiang Z, Zhao F, Xi B, Zhang X, Fu H, Zhou K. 2015. Platycodin-D Induced Autophagy in Non-Small Cell Lung Cancer Cells via PI3K/Akt/mTOR and MAPK Signaling Pathways. J Cancer. 6(7):623-631.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) License. See http://ivyspring.com/terms for full terms and conditions.