Journal of Cancer 3.2
Impact Factor
ISSN: 1837-9664
Impact Factor
ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2015; 6(5):470-476. doi:10.7150/jca.11494 This issue Cite
Research Paper
Role of Mitochondrial Network Stabilisation by a Human Small Heat Shock Protein in Tumour Malignancy
Zsuzsanna Turi1*, Eniko Hocsak1*, Boglarka Racz1, Aliz Szabo1, Andras Balogh2, Balazs Sumegi1,3,4, Ferenc Gallyas Jr.1,3,4 ![]()
1. Department of Biochemistry and Medical Chemistry, University of Pécs Medical School, 12 Szigeti St., H-7624 Pécs, Hungary;
2. Department of Medical Biology, University of Pécs Medical School, 12 Szigeti St., H-7624 Pécs, Hungary;
3. MTA-PTE Nuclear-Mitochondrial Interactions Research Group, University of Pécs Medical School, 12 Szigeti St., H-7624 Pécs, Hungary;
4. Szentagothai Research Centre, 34 Ifjusag St., H-7624 Pécs, Hungary.
* These authors made equal contributions to this work.
Received 2015-1-5; Accepted 2015-2-14; Published 2015-3-25
Citation:
Turi Z, Hocsak E, Racz B, Szabo A, Balogh A, Sumegi B, Gallyas F Jr.. Role of Mitochondrial Network Stabilisation by a Human Small Heat Shock Protein in Tumour Malignancy. J Cancer 2015; 6(5):470-476. doi:10.7150/jca.11494. https://www.jcancer.org/v06p0470.htm
Other stylesAbstract
Previously, we found that the unconventional small human heat-shock protein HSPB11 inhibits cell death by HSP90 mediated cholesterol-rich membrane microdomain dependent activation of phosphatidylinositol-3 kinase/protein kinase B pathway and by stabilising the mitochondrial membrane systems. Also, progressive cytoplasmic expression of HSPB11 correlated with brain tumor malignancy. In the present study we investigated how cytoplasmic abundance of HSPB11 augments tumor malignancy. We up- and downregulated the cytoplasmic level of HSPB11 before paclitaxel exposure in NIH3T3 and HeLa cells, which normally express low and high levels, respectively, of the HSPB11 protein. We examined the paclitaxel-mediated induction of cell death, mitochondrial fission, HSPB11 mitochondrial translocation and inhibitory phosphorylation of dynamin-like protein-1 (DLP1). We found that increasing cytoplasmic abundance of HSPB11 in NIH3T3 cells protected against paclitaxel-induced apoptosis, while suppressing HSPB11 in HeLa cells sensitised the cells toward paclitaxel. Also, paclitaxel enhanced mitochondrial translocation of HSPB11 in wild type HeLa but not in NIH3T3 cells. More importantly, increased cytoplasmic level of HSPB11 in NIH3T3 cells enhanced the inhibitory phosphorylation of DLP1 and attenuated paclitaxel-induced mitochondrial fission. All these results suggest that increased cytoplasmic abundance of HSPB11 augments inhibitory phosphorylation of DLP1 thereby reduces mitochondrial fission that eventually leads to decreased apoptosis. This novel mechanism may explain the resistance to apoptosis and increased malignancy of HSPB11-overexpressing tumours. The clinical significance of this mechanism has already been highlighted by the finding that the kinase inhibitor tyrphostin A9 induces cancer cell death by DLP1-mediated mitochondrial fragmentation.
Keywords: sHSP, HSPB11, cytoprotection, HeLa, mitochondrial fission, DLP1, apoptosis, tumour malignancy.
Introduction
Small heat shock proteins (sHSPs) promote cell survival by preventing the aggregation of nascent, stress-accumulated, misfolded proteins and directly interacting with various components of the apoptotic machinery [1,2]. The overexpression of sHSPs may increase malignancy and cytostatic tumour resistance. The most extensively characterised sHSP family members, HSPB1 and HSPB5, were found to modulate metastatic potential and tumour progression [3]. We established a positive correlation between cytoplasmic HSPB11 expression and histological grade of brain tumours [4]. Increased expression of sHSPs was proposed as a diagnostic tool, and mechanisms of the regulation of sHSP expression or oligomerisation were suggested as therapeutic targets for clinical tumour management [1, 3].
Mitochondria undergo functional and ultrastructural remodelling during cancer development, which involves the cristae structure, the function of the energy-generating oxidative phosphorylation system; proteins associated with apoptosis regulation and the protein composition and association properties of membrane systems and mitochondrial network fragmentation [5]. The remodelling process affects hypoxia tolerance, sensitivity to apoptosis-inducing conditions [6], chemoresistance and metastatic potential [7]. Recent reports have described dramatic alterations in mitochondrial morphology during the early stages of apoptotic cell death, one of which is mitochondrial fission and the subsequent release of pro-apoptotic mitochondrial intermembrane proteins, which results in mitochondrial fragmentation [5,7]. The fragmentation of the mitochondrial network occurs when an abnormal mitochondrial inner membrane potential develops in response to the impairment of oxidative phosphorylation [6]. Mitochondrial fragmentation, however, does not necessarily lead to apoptosis [5,6]. The mitochondrial fission pathway involves at least two proteins: a cytosolic GTPase, dynamin-like protein 1 (DLP1), and a mitochondrial outer membrane protein, fission 1 (FIS1) [7]. FIS1 is considered the limiting factor in the fission process because it recruits DLP1 to punctate structures on the mitochondrial surface [7]. DLP1 is regulated by phosphorylation-dephosphorylation. The phosphorylation of Ser637 of DLP1 by cAMP-dependent protein kinase (PKA) impairs the GTPase activity of DLP1, thereby preventing mitochondrial fission. PKA prevents mitochondrial fission without interfering with the GTPase activity of DLP1 by phosphorylating Ser656, which most likely impedes oligomerisation [8].
To further elucidate the connection between HSPB11 expression and tumour malignancy, we investigated the association between HSPB11 and mitochondria under stress conditions, and we analysed the effects of HSPB11 on mitochondrial fission and DLP1 phosphorylation in cell lines with different levels of cytosolic HSPB11.
Materials and Methods
Materials
Paclitaxel (Taxol) was obtained from ICN Biochemicals Inc., and MitoTracker Red CMXRos was from Invitrogen. Protease and phosphatase inhibitor cocktails, propidium iodide (PI), anti-actin rabbit antibody, anti-histone H3 rabbit antibody, anti-mouse IgG, anti-rabbit IgG and all cell culture chemicals were purchased from Sigma-Aldrich Ltd. The mammalian expression vectors pcDNA3.1 and pEGFP-C1 were from Invitrogen and BD Biosciences, respectively. The anti-glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and anti-phospho-DLP1 (Ser637) rabbit antibodies were from Cell Signaling, and the rabbit IgG and anti-DLP1 (C5) mouse monoclonal antibodies were from Santa Cruz Biotechnology. The anti-pyruvate dehydrogenase complex (PDC)-1α and anti-HSPB11 polyclonal antibodies were custom-made.
Cell lines
NIH3T3 and HeLa cells were purchased from the American Type Culture Collection. The culture and harvesting of cells, isolation of cytosolic and mitochondrial fractions and immunoblotting were performed as previously described [9].
Overexpression and silencing of HSPB11
To overexpress HSPB11 or green fluorescent protein (GFP)-tagged HSPB11, the entire HSPB11 coding sequence was PCR-amplified and inserted into the pcDNA3.1 (Invitrogen) or pEGFP-C1 (BD Biosciences) mammalian expression vector, respectively. To silence HSPB11, the BLOCK-iT RNAi TOPO Transcription Kit and the BLOCK-iT Dicer RNAi Transfection Kit were used according to the manufacturer's protocol (Invitrogen). Transient transfection was performed in Opti-MEM I medium using Lipofectamine 2000 (Invitrogen).
Flow cytometry analysis
After paclitaxel-treatment, the cells were rinsed and harvested. The Annexin-V-FLUOS Staining Kit (Roche Applied Science) was used according to the manufacturer's protocol, and the cells were analysed using flow cytometry in a BD FACSCalibur flow cytometer (BD Biosciences). Quadrant dot plots were created using CellQuest software (BD Biosciences), and the numbers of live, necrotic and apoptotic cells were expressed as a percentage of the total number of stained cells. All the experiments were repeated three times.
Confocal microscopy
Samples were visualised using an Olympus FluoView 1000 confocal laser scanning microscope. Excitation by a multiline argon-ion laser at 488 nm and green helium-neon laser at 543 nm was used to visualise GFP-tagged HSPB11 and MitoTracker Red, respectively, at 10 µs/pixel in photon-counting and sequential mode. The field of interest was scanned in XY or XYZ mode to observe a single layer or the total thickness of the cells at a 1.5-µm layer distance, respectively, and images were acquired at 1024 x 1024 pixels/layer. All the experiments were repeated three times.
Assessing mitochondrial fragmentation
For assessing mitochondrial fragmentation, we determined mitochondrial fragmentation count (MFC) exactly as it was described by Rehman et al. [10] from the MitoTracker Red images. Briefly, for each treatment group, images of at least 25 randomly selected cells were background subtracted, filtered (median), thresholded, and binarised using ImageJ (US NIH) software. MFC was calculated by normalising the number of continuous mitochondrial structures to the total mitochondrial pixel count [10].
Statistical analysis
The mean ± the standard error of the mean (SEM) values were analysed using a one-way or two-way analysis of variance followed by Tukey's post-hoc test. When the F-test indicated unequal variances, the Kruskal-Wallis test was performed.
Results
Cytoprotection by HSPB11 is primarily achieved by inhibiting apoptosis
We increased the cytoplasmic abundance of HSPB11 by overexpressing it in NIH3T3 fibroblasts that normally express the protein at low extent. After double-staining the cells with fluorescein-labelled Annexin-V and propidium iodide, we determined the type of death induced by a 3 h exposure to 150 nM paclitaxel using flow cytometry. This approach is widely used to detect apoptosis and necrosis. Annexin-V binds to phosphatidylserine residues that appear on the outer surface of the cell membrane as an early indicator of apoptosis, whereas the entry of propidium iodide into the cell indicates a loss of membrane integrity and therefore necrosis. As demonstrated in Figure 1A, the transfection of our construct effectively increased the steady-state level of HSPB11. In sham-transfected NIH3T3 cells, paclitaxel treatment induced apoptotic death in 29.0±2.8% of the cells and necrotic death in 21.0±2.1% of them (Fig. 1B and C). The levels of paclitaxel-induced apoptosis and necrosis decreased to 12.3±1.4% (p<0.05) and 17.6±1.5% (not significant), respectively, in HSPB11-overexpressing NIH3T3 cells (Fig. 1B and C), confirming that HSPB11 reduced apoptosis.
Figure 1
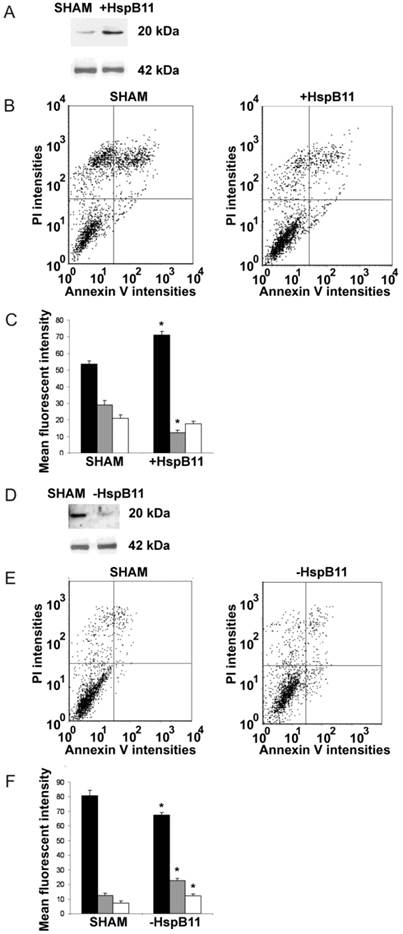
Effect of HspB11 on paclitaxel-induced cell death. NIH3T3 fibroblasts (A, B, C) and HeLa cervical carcinoma cells (D, E, F) were transfected with empty (SHAM), HspB11-expressing (+HspB11) or HspB11-silencing (-HspB11) plasmids. The expression of HspB11 (A, D) was determined using immunoblotting with a custom-made polyclonal anti-HspB11 primary antibody. Actin was used as a loading control. After transfection, the cells were exposed to 150 nM paclitaxel for 3 h and double-stained with fluorescein-conjugated Annexin V and PI, and necrosis and apoptosis were assessed using flow cytometry. Representative dot-plots (B, E) and bar diagrams (C, F) of three independent experiments are presented. The horizontal and vertical axes of the dot-plots represent the Annexin V and PI staining intensities, respectively. Lower left quadrant, living cells; lower right quadrant, early apoptotic cells; upper left quadrant, necrotic cells; upper right quadrant, late apoptotic cells. The bar diagrams represent living (black bars), apoptotic (grey bars) and necrotic (white bars) cells. The values are the mean ± SEM, and significant differences compared with the appropriate SHAM values are indicated as * (p<0.05).
As an opposite approach, we silenced HSPB11 using the Dicer-substrate siRNA (DsiRNA) technique in HeLa cells (Fig. 1D), which express high levels of endogenous HSPB11. Compared with sham-transfected wild-type cells, HSPB11 silencing sensitised the cells to paclitaxel-induced apoptotic and necrotic cell death (Fig. 1E and F). We found a significant increase in apoptosis compared with necrosis in the HSPB11-silenced HeLa cells, which supported our findings in the HSPB11-overexpressing NIH3T3 cells.
HSPB11 protects against mitochondrial fission
Mitochondrial fission, one of the earliest events in the apoptotic process, precedes the release of pro-apoptotic mitochondrial intermembrane proteins. To elucidate how HSPB11 affects mitochondrial fission, we investigated the effect of the cytoplasmic level of HSPB11 on paclitaxel-induced mitochondrial fission. We transiently transfected NIH3T3 cells with the HSPB11-GFP fusion protein-overexpressing plasmid, and then the cells were exposed or not to 150 nM paclitaxel for 3 h. We monitored the cells using laser scanning confocal microscopy after MitoTracker Red loading. Floating cells killed by the paclitaxel treatment were discarded by washing before imaging. Unlike the >80% efficacy of our previous experiments, only 46±11% of the cells were successfully transfected with this plasmid, enabling the observation of identically treated and processed wild-type and HSPB11-GFP-overexpressing cells in the same microscopic field. As shown in the upper panels of Figure 2A, the wild-type cells exhibited a branching tubular mitochondrial network regardless of the cytoplasmic abundance of HSPB11-GFP fusion protein. Upon paclitaxel treatment, approximately 80% of the wild-type cells underwent mitochondrial fission, resulting in a punctate distribution of evenly dispersed clusters of mitochondria and a reduction of the red fluorescence intensity. The latter result indicated an impaired mitochondrial membrane potential. In contrast, paclitaxel-treated, HSPB11-GFP fusion protein-expressing NIH3T3 cells, which can be identified by their green-yellow fluorescence in the right panels, retained the morphology and red fluorescence intensity of the wild-type cells (lower panels of Fig. 2A). The mitochondrial morphology of the paclitaxel-treated NIH3T3 cells transfected with GFP alone was identical to that of the wild-type NIH3T3 cells (data not shown), indicating that the GFP portion of the fusion protein was not involved in the protection against paclitaxel-induced mitochondrial fission.
We assessed the level of mitochondrial fragmentation quantitatively by determining MFC [10] values of randomly selected 25 cells for each of the different groups (untreated―untransfected, untreated―HSPB11-GFP-transfected, paclitaxel-treated―untransfected and paclitaxel-treated― HSPB11-GFP-transfected) using the MitoTracker Red fluorescent images of the aforementioned experiment. We found significantly increased fragmentation only in the paclitaxel-treated―untransfected group (Fig. 2B).
Figure 2
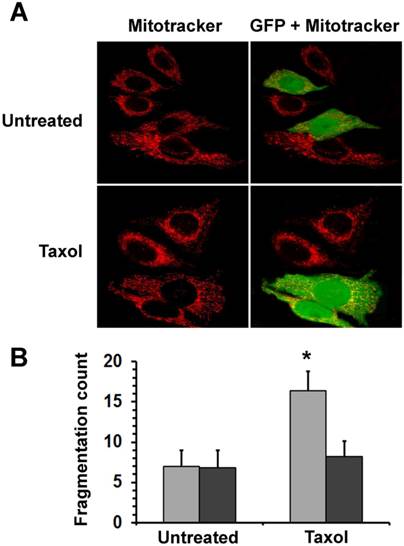
Effects of HspB11 and paclitaxel on mitochondrial fission. NIH3T3 fibroblasts were transiently transfected with a plasmid expressing a HspB11-GFP fusion protein and were exposed to 150 nM paclitaxel (Taxol) or not exposed (Untreated) for 3 h. After loading of the red fluorescent mitochondrial dye MitoTracker Red, mitochondria and GFP were visualised using laser scanning confocal microscopy. Representative images (A) of three independent experiments are presented. The left (red channel) and right (merged red and green channels) panels depict the same microscopic field. The HspB11-overexpressing cells are green-yellow in the right panels, whereas the wild-type cells are red. Mitochondrial fragmentation counts (B) were determined from the MitoTracker Red images, and mean ± SEM fragmentation values of randomly selected 25 cells (per group) are presented as a bar diagram. Significant differences between SHAM (light bars) and transfected (dark bars) are indicated as * (p<0.05).
Paclitaxel increases the mitochondrial localisation of HSPB11 in HeLa cells
Previously, we found that recombinant HSPB11 protected isolated mitochondria from high-calcium-induced depolarisation [11], suggesting a direct interaction between HSPB11 and mitochondria. To verify whether a similar direct interaction was responsible for the fission resistance, we investigated the mitochondrial localisation of HSPB11 after paclitaxel treatment. HeLa and NIH3T3 cells were exposed or not to 150 nM paclitaxel for 3 h. After isolating the cytosolic and mitochondrial fractions, we determined the level of HSPB11 in the fractions using immunoblotting. The purity of the intracellular fractions was ensured by the use of appropriate markers, which also served as loading controls. Paclitaxel treatment slightly (about 20%) enhanced the HSPB11 level in all the fractions except for the mitochondrial fraction of HeLa cells, in which the increase in HSPB11 was significantly enhanced (37%, p<0.05; Fig. 3A and B).
Figure 3
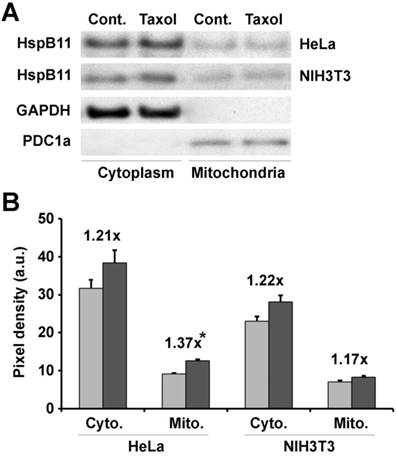
Effects of paclitaxel on HspB11 translocation. After subcellular fractionation, the cytoplasmic (Cyto.) and mitochondrial (Mito.) levels of HspB11 in paclitaxel-treated (dark bars) and untreated (Cont. and light bars) HeLa and NIH3T3 cells were assessed using immunoblotting with a custom-made, polyclonal anti-HspB11 primary antibody. Representative blots (A) and bar diagrams (B) of three independent experiments are presented. In addition to the anti-HspB11 antibody, subcellular fractions were also stained with anti-glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and anti-pyruvate dehydrogenase complex-1α (PDC1a) primary antibodies to indicate the purity of the subcellular fraction and serve as loading controls. The values represent the mean ± SEM, the Taxol per Cont. values are indicated above the bars, and significant differences from the paclitaxel-induced increase in the cytoplasmic level of HspB11 in HeLa cells are indicated as * (p<0.05).
HSPB11 induces inhibitory phosphorylation and protects against the paclitaxel-induced dephosphorylation of DLP1
Because the inhibitory phosphorylation of DLP1 is considered a major regulator of mitochondrial fission [8], we investigated the influence of the cytoplasmic level of HSPB11 on this process. We used the same experimental approach as for studying cell death and examined the expression level and phosphorylation state of DLP1 using immunoblotting. We found that neither the cytoplasmic level of HSPB11 nor paclitaxel treatment affected the steady-state expression level of DLP1. In contrast, the inhibitory phosphorylation of DLP1 was reduced by approximately half when HSPB11 was silenced in HeLa cells, and it was doubled when HSPB11 was overexpressed in NIH3T3 cells. As expected, paclitaxel decreased DLP1 phosphorylation, regardless of the cytoplasmic level of HSPB11 or the cell type (Fig. 4).
Figure 4
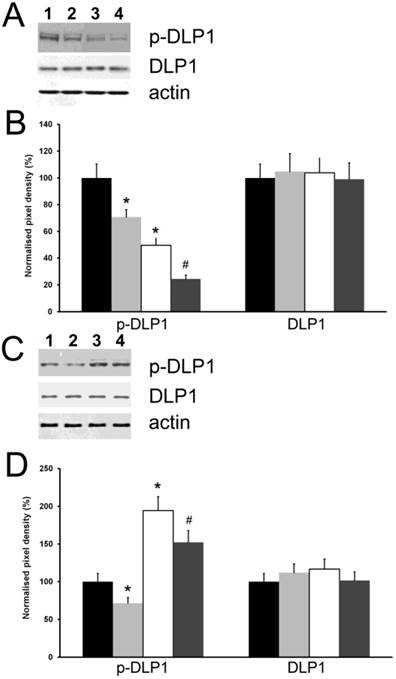
Effect of HspB11 on paclitaxel-induced dephosphorylation of DLP1. HeLa cervical carcinoma cells (A, B) and NIH3T3 fibroblasts (C, D) were transfected with empty, HspB11-silencing (A, B) and HspB11-expressing plasmids (C, D) and were then exposed or not to 150 nM paclitaxel for 3 h. The expression and phosphorylation state of DLP1 were assessed using immunoblotting with anti-DLP1 and phospho(Ser637)-specific anti-DLP1 primary antibodies, respectively, in four experimental groups: sham-transfected-untreated (1 and black bars), sham-transfected-paclitaxel-treated (2 and light grey bars), transfected-untreated (3 and white bars) and transfected-paclitaxel-treated (4 and dark grey bars). Representative blots (A, C) and bar diagrams (B, D) from three independent experiments are presented. Actin was used as a loading control, and the pixel densities were normalised to the appropriate actin value. The values represent the mean ± SEM; significant differences from the sham-transfected-untreated group are indicated as * (p<0.05). Significant differences from the transfected-untreated groups are indicated as # (p<0.05).
Discussion
In a previous study of 91 human samples, we found a correlation between the cytoplasmic expression of HSPB11 and the clinical grade of brain tumours [4]. In an effort to explain this correlation, we up- and downregulated the cytoplasmic expression of HSPB11 in vitro in the present work and studied the paclitaxel-induced apoptosis under these conditions. Using a brief exposure to paclitaxel known to cause moderate necrotic and apoptotic cell death [12], we observed that HSPB11 overexpression protected NIH3T3 cells against paclitaxel by means of decreased apoptosis. In contrast, increased levels of necrosis and apoptosis were equally responsible for the observed vulnerability to paclitaxel of HSPB11-gene-silenced HeLa cells. We assume that the difference between the cell lines was because, unlike HeLa cells, NIH3T3 cells have not evolved compensatory mechanisms that block or mask the effect of HSPB11.
Mitochondria play pivotal roles in necrotic and apoptotic cell death, and they become damage-prone upon fragmentation [13]. Therefore, we studied the effect of HSPB11 on paclitaxel-induced mitochondrial fission. We overexpressed an HSPB11-GFP fusion protein in the NIH3T3 cell line. The GFP tag enabled us to distinguish cells expressing high levels of HSPB11 from their wild-type counterparts, and we compared the paclitaxel-induced changes in the mitochondrial networks of the two cell types. We found that an increased cytoplasmic abundance of HSPB11 stabilised the mitochondrial network and protected against paclitaxel-induced mitochondrial fission by increasing the inhibitory phosphorylation of DLP1. Furthermore, the paclitaxel-stimulated mitochondrial translocation of HSPB11 occurred in HeLa cells, but not in NIH3T3 cells, indicating the necessity of a high HSPB11 cytoplasmic abundance in this process. Previously, we found that heme-binding protein 2/SOUL, a B-cell lymphoma 2 homology domain (BH)3-only protein that facilitates the mitochondrial permeability transition and mainly localises to the cytoplasm, translocated to the mitochondria upon stimulation [14]. In the present study, paclitaxel induced the mitochondrial accumulation of HSPB11 in HeLa cells to approximately the same extent as previously found for SOUL.
All of these findings suggest the following model for the connection between HSPB11 expression and tumour malignancy. In normal cells, HSPB11 is mainly localised to the nucleus. The increased HSPB11 expression associated with the higher protein turnover rate in tumour cells results in increased cytoplasmic localisation of the protein. Under stress conditions, HSPB11 can translocate to the mitochondria and increase the inhibitory phosphorylation of DLP1, thereby stabilising the mitochondrial network, which increases the cell's resistance to apoptosis, cytostatic drugs, and consequently, the overall malignancy of the tumour cells.
Conclusions
All the findings presented indicate that increased cytoplasmic abundance of HSPB11 augments inhibitory phosphorylation of DLP1 thereby reduces mitochondrial fission that eventually leads to decreased apoptosis. To the best of our knowledge, this is the first study describing an effect of an sHSP on mitochondrial fission and DLP1 phosphorylation. Whether this effect is a common characteristic of sHSPs or unique to HSPB11 remains unclear. It is noteworthy that the receptor tyrosine kinase inhibitor tyrphostin A9 induces cancer cell death by DLP1-mediated mitochondrial fragmentation [15] that highlights the clinical significance of the aforementioned mechanism.
Abbreviations
HSP: heat shock protein; DLP1: dynamin-like protein-1; sHSP: small heat shock protein; FIS1: fission (protein) 1; PKA: cAMP-dependent protein kinase; PI: propidium iodide; GAPDH: glyceraldehyde 3-phosphate dehydrogenase; PDC: pyruvate dehydrogenase complex; GFP: green fluorescent protein; MFC: mitochondrial fragmentation count; SEM: standard error of the mean; BH3: B-cell lymphoma 2 homology domain 3.
Acknowledgements
This research was realized in the frames of TÁMOP 4.2.4. A/2-11-1-2012-0001 “National Excellence Program - Elaborating and operating an inland student and researcher personal support system” The project was subsidized by the European Union and co-financed by the European Social Fund (FG). This work was also supported by Hungarian grants OTKA NN109841 (FG), K104220 (BS), PD105589 (BR), 34039/KA-OTKA/11-17 (EH) and GVOP-321-2004-04-0172/3.0 (AB).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Sreedhar AS, Csermely P. Heat shock proteins in the regulation of apoptosis: new strategies in tumor therapy: a comprehensive review. Pharmacol Ther. 2004;101:227-57
2. Ojha J, Masilamoni G, Dunlap D. et al. Sequestration of toxic oligomers by HspB1 as a cytoprotective mechanism. Mol Cell Biol. 2011;31:3146-57
3. Arrigo AP, Simon S, Gibert B. et al. Hsp27 (HspB1) and alphaB-crystallin (HspB5) as therapeutic targets. FEBS Lett. 2007;581:3665-74
4. Pozsgai E, Gomori E, Szigeti A. et al. Correlation between the progressive cytoplasmic expression of a novel small heat shock protein (Hsp16.2) and malignancy in brain tumors. BMC Cancer. 2007;7:233-9
5. Arismendi-Morillo G. Electron microscopy morphology of the mitochondrial network in human cancer. Int J Biochem Cell Biol. 2009;41:2062-68
6. Benard G, Bellance N, James D. et al. Mitochondrial bioenergetics and structural network organization. J Cell Sci. 2007;12:838-48
7. Alirol E, Martinou JC. Mitochondria and cancer: is there a morphological connection? Oncogene. 2006;25:4706-16
8. Cribbs JT, Strack S. Reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. EMBO Rep. 2007;8:939-44
9. Tapodi A, Debreceni B, Hanto K. et al. Pivotal role of Akt activation in mitochondrial protection and cell survival by poly(ADP-ribose)polymerase-1 inhibition in oxidative stress. J Biol Chem. 2005;280:35767-75
10. Rehman J1, Zhang HJ, Toth PT. et al. Inhibition of mitochondrial fission prevents cell cycle progression in lung cancer. FASEB J. 2012;26:2175-86
11. Bellyei S, Szigeti A, Boronkai A. et al. Inhibition of cell death by a novel 16.2 kD heat shock protein predominantly via Hsp90 mediated lipid rafts stabilization and Akt activation pathway. Apoptosis. 2007;12:97-112
12. Varbiro G, Veres B, Gallyas FJr. et al. Direct effect of taxol on free radical formation and mitochondrial permeability transition. Free Rad Biol Med. 2001;31:548-58
13. Dhingra R, Kirshenbaum LA. Regulation of mitochondrial dynamics and cell fate. Circ.J. 2014;78:803-10
14. Szigeti A, Hocsak E, Rapolti E. et al. Facilitation of mitochondrial outer and inner membrane permeabilization and cell death in oxidative stress by a novel Bcl-2 homology 3 domain protein. J Biol Chem. 2010;285:2140-51
15. Park SJ, Park YJ, Shin JH. et al. A receptor tyrosine kinase inhibitor, Tyrphostin A9 induces cancer cell death through Drp1 dependent mitochondria fragmentation. Biochem Biophys Res Commun. 2011;408:465-70
Author contact
![]() Corresponding author: Ferenc Gallyas Jr., PhD, DSc, Department of Biochemistry and Medical Chemistry, University of Pécs, 12 Szigeti St., Pécs H-7624, Hungary. Tel: +36-72-536-279, Fax: 36-72-536-277, E-mail: ferenc.gallyaspte.hu
Corresponding author: Ferenc Gallyas Jr., PhD, DSc, Department of Biochemistry and Medical Chemistry, University of Pécs, 12 Szigeti St., Pécs H-7624, Hungary. Tel: +36-72-536-279, Fax: 36-72-536-277, E-mail: ferenc.gallyaspte.hu
Citation styles
APA
Turi, Z., Hocsak, E., Racz, B., Szabo, A., Balogh, A., Sumegi, B., Gallyas, F. Jr. (2015). Role of Mitochondrial Network Stabilisation by a Human Small Heat Shock Protein in Tumour Malignancy. Journal of Cancer, 6(5), 470-476. https://doi.org/10.7150/jca.11494.
ACS
Turi, Z.; Hocsak, E.; Racz, B.; Szabo, A.; Balogh, A.; Sumegi, B.; Gallyas, F. Jr. Role of Mitochondrial Network Stabilisation by a Human Small Heat Shock Protein in Tumour Malignancy. J. Cancer 2015, 6 (5), 470-476. DOI: 10.7150/jca.11494.
NLM
Turi Z, Hocsak E, Racz B, Szabo A, Balogh A, Sumegi B, Gallyas F Jr.. Role of Mitochondrial Network Stabilisation by a Human Small Heat Shock Protein in Tumour Malignancy. J Cancer 2015; 6(5):470-476. doi:10.7150/jca.11494. https://www.jcancer.org/v06p0470.htm
CSE
Turi Z, Hocsak E, Racz B, Szabo A, Balogh A, Sumegi B, Gallyas F Jr.. 2015. Role of Mitochondrial Network Stabilisation by a Human Small Heat Shock Protein in Tumour Malignancy. J Cancer. 6(5):470-476.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) License. See http://ivyspring.com/terms for full terms and conditions.