Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2015; 6(4):319-326. doi:10.7150/jca.10733 This issue Cite
Research Paper
YSY01A, a Novel Proteasome Inhibitor, Induces Cell Cycle Arrest on G2 Phase in MCF-7 Cells via ERα and PI3K/Akt Pathways
Bingjie Xue1, Wei Huang1, Xia Yuan1, Bo Xu2, Yaxin Lou3, Quan Zhou1, Fuxiang Ran1, Zemei Ge4, Runtao Li4 ![]() , Jingrong Cui1
, Jingrong Cui1 ![]()
1. State Key Laboratory of Natural and Biomimetic Drugs, School of Pharmaceutical Sciences, Peking University, 100083, Beijing, China;
2. Instrumental Analysis Center of State Key Laboratory of Natural and Biomimetic Drugs, School of Pharmaceutical Sciences, Peking University, 100083, Beijing, China;
3. Lab of Proteomics Medical and Healthy Analytical Center, Peking University, Beijing, China;
4. Peking University School of Pharmaceutical Sciences Department of Medicinal Chemistry, Beijing, China
Received 2014-10-5; Accepted 2014-11-15; Published 2015-2-6
Abstract
Given that the proteasome is essential for multiple cellular processes by degrading diverse regulatory proteins, inhibition of the proteasome has emerged as an attractive target for anti-cancer therapy. YSY01A is a novel small molecule compound targeting the proteasome. The compound was found to suppress viability of MCF-7 cells and cause limited cell membrane damage as determined by sulforhodamine B assay (SRB) and CytoTox 96® non-radioactive cytotoxicity assay. High-content screening (HCS) further shows that YSY01A treatment induces cell cycle arrest on G2 phase within 24 hrs. Label-free quantitative proteomics (LFQP), which allows extensive comparison of cellular responses following YSY01A treatment, suggests that various regulatory proteins including cell cycle associated proteins and PI3K/Akt pathway may be affected. Furthermore, YSY01A increases p-CDC-2, p-FOXO3a, p53, p21Cip1 and p27Kip1 but decreases p-Akt, p-ERα as confirmed by Western blotting. Therefore, YSY01A represents a potential therapeutic for breast cancer MCF-7 by inducing G2 phase arrest via ERα and PI3K/Akt pathways.
Keywords: YSY01A, PS341, MCF-7, High-content screening, Label-free quantitative proteomics, ERα, PI3K/Akt pathways
Introduction
The ubiquitin proteasome system (UPS) is crucial for the turnover of proteins by controlling cell cycle, programmed cell death, cell proliferation, survival, adhesion and differentiation1. UPS dysregulation is implicated in the underlying molecular pathology of a variety of diseases, including cancer2. Ubiquitin and various proteasome have thus become an attractive therapeutic target for treatment of malignancies. In the UPS, several ubiquitin ligases, such as RNF123, have been shown to bind directly to estrogen receptor-α (ERα) and regulate its activity. Besides, a number of studies have suggested that proteasome inhibition alters ERα-dependent gene transcription via diverse effects and mechanisms4,5-8. For example, PS341 have been reported that inhibited ERα due to direct transcriptional inhibition and loss of RNA polymerase II recruitment on the ERα gene promote7. The ERα, expressed in over two thirds of breast cancers, plays an essential role on tumor growth9-10, and blockade of the estrogen action is the mainstay of treatment of ER-positive breast cancer. However, activation of the phosphoinositide 3-kinase (PI3K) pathway occurs frequently in breast cancer. The over-activation of PI3K pathway promotes resistance to the selective estrogen receptor modulators (SERMs) tamoxifen or aromatase inhibitor letrozole which are the first-line treatment for patients with ERα-positive breast cancer11. The molecular mechanism reveals that ERα and the PI3K/Akt pathways form a positive feedback and their cross-talk has also been demonstrated in cancer cells. As the downstream target of PI3K/Akt signaling pathway, the kinase p70S6K can activate ERα by phosphorylation at Ser167 residue. Subsequently, activated ERα acts on PI3K, resulting in a positive feedback loop12,5. Inhibition of PI3K pathway and ERα by proteasome inhibitors therefore represents a potentially attractive strategy for the treatment of breast cancer13,14-15.
Proteasome inhibitor, as a new anticancer agent, is promising for anticancer therapy that benefit patient with multiple myeloma and non-hodgkin's lymphoma. Bortezomib (also called PS341) have been approved for the treatment of multiple myeloma or relapsed/refractory mantle cell lymphoma16-17. These studies also reported that multiple myeloma cell lines that were previously resistant to melphalan, doxorubicin, dexamethasone, or mitoxantrone were sensitized up to 1,000,000-fold by prior exposure to subtoxic concentrations of bortezomib are in clinical development because of their anti-neoplastic and anti-inflammatory18-19. Bortezomib blocks TNF-α induced NF-κB activation in a dose- and time-dependent manner in multiplemyeloma
cells through degradation of IκBα20. Carfilzomib, the second generation proteasome inhibitor, is a more selective proteasome inhibitor with more mild toxicity compared to bortezomib and is also able to overcome resistance to chemotherapeutic agents21. Besides, there are some proteasome inhibitors are under investigation, such as Marizomib (NPI-0052), Ixazomib (MLN9708), which are safety and efficacy in some patients from both phase I and II trials22. However, their antitumor activity remains unsatisfactory, especially in most solid tumors such as breast cacner23-24. Herein, there is considerable interest in seeking a proteasome inhibitor for the treatment of solid tumor malignancies.
Recently, our collaborator, Dr. RT Li, identified a novel proteasome inhibitor, YSY01A, we have demonstrated that YSY01A has less toxicity to livers, kidneys and intestines of nude mice and ICR mice compared with PS341 (data to be published). In this study, we further investigate inhibitory effect of YSY01A on tumor cell viability and find that the compound induces cell cycle arrest in MCF-7 cells. Together, we conclude that YSY01A promises a novel probe for development of proteasome inhibitors and a potential therapeutic for breast cancer treatment.
Materials and methods
Cell Culture
Human breast cancer cells MCF-7 were maintained as previously described25. For all experiments, cells were cultured in RPMI-1640 medium (Macgene, China) supplemented with 10% fetal bovine serum, 100 U/ml penicillin, and 100 μg/ml streptomycin (Macgene, China) in a 37°C humidified atmosphere containing 5% CO2 (Heraeus, Germany).
Cell Viability Assay
Cell viability and cytotoxicity of YSY01A to MCF-7 cells was determined using sulforhodamine B (SRB) colorimetric assay and CytoTox96® non-radioactive cytotoxicity assay. Briefly, 4,000 cells/well of MCF-7 cells were plated in a 96-well plate overnight. Cells were then treated with vehicle control (PBS), YSY01A (20, 40, 80 nM) or PS341 (40 nM) for 12, 24 and 48 hrs (SRB assay) or 24, 48 and 96 hrs (CytoTox96® assay), respectively. SRB assay was carried out as previously described.25 For CytoTox96® assay, cells were firstly lyzed following the manufacturer's instructions for determination of the maximum LDH release. On the other side, released LDH in culture medium of parallel groups was measured after brief spin down and harvest for detection of cell damage following treatments. The percentage of cellular injury was calculated using the formula: % cytoxicity = experimental LDH released / maximum LDH release×100 and normalized to vehicle control26.
Image Based Quantitative Analysis to Determine Cell Cycle Distribution by HCS
Click-iT® Alexa Fluor® 488 TUNEL imaging assay (Invitrogen, USA) was used to determine the effects of YSY01A on cell cycle. MCF-7 cells were seeded overnight in a 96 well imaging plate followed by treatment with 20, 40 and 80 nM YSY01A for 24 hrs, and then fixed with EdU (5-ethynyl-2'-deoxyuridine) for 1 hr and labeled with Alexa Fluor® 488 probe or labeled with antibody against phospho-histone 3 (p-H3). Cells were also co-stained with Hoechst 33342 dye to determine the DNA content based on the nuclear intensity and finally analyzed by high-content screening system equipped with Columbus analysis tool (Cellomics, USA). Cell cycle distribution was defined to S phase by incorporation of the thymidine analog, EdU, and M phase by immunostaining for the mitotic marker, p-H3, while the remaining unlabeled cells were identified to undergo G1 or G2 phases27.
2D LC-MS/MS Label-free Quantitative Proteomic Analysis
2D LC-MS/MS label-free quantitative proteomic analysis was used to investigate the molecular profiles of YSY01A in MCF-7 cells. To prepare cell culture samples, MCF-7 cells were exposed to vehicle control (PBS), YSY01A or PS341 for 24 hrs and lysed in ice-cold lysis buffer (8 M Urea, 75 mM NaCl, 50 mM Tris, protease inhibitor cocktail, pH 8.2) followed by sonication and high-speed centrifugation at 14,000 rpm for 30 mins. The resulting supernatants were harvested and their protein concentrations were measured using bicinchoninic acid protein assay (Dingguo Changsheng Biotechnology, China). Prior to proteomic analysis, protein samples were reduced by incubation with DTT at a final concentration of 5 mM for 25 mins at 56 °C and then alkylated by 14mM iodoacetamide for 30 mins in the dark. Unreacted iodoacetamide was quenched by adding 0.5 M DTT and incubated for 15 mins at room temperature in the dark, and the protein mixture was subsequently diluted 1:5 in 25 mM Tris-HCl, pH 8.2, to reduce the concentration of urea to 1.6M. CaCl2 was added to a final concentration of 1 mM with addition of trypsin at 37 °C overnight. The digestion was stopped by acidification with 0.4% (vol/vol) trifluoroacetic acid followed by pH adjustment to 2.0 and centrifugation at 2,500g for 10 mins at room temperature. 200 μg of peptides from each sample were separated into 15 fractions by strong anion exchange as described previously28. For 2D LC-MS/MS analysis, the eluted peptides were concentrated and purified and separated by reverse-phase chromatography (XbridgeTM C18 3.5um, 4.6x20mm column, and XbridgeTM BEH130 C18 3.5um,2.1x150mm column, Waters, Ireland). A nanoflow high performance liquid chromatography (HPLC) system (EASY-column,10 cm,ID 75um,3um, Thermo Fisher, USA) equipped was used to analysis, followed by redissolving in formic acid, and loading onto the column (EASY-column, 2 cm, ID 100um, 5um,C18, Thermo Fisher, USA). The 15 high-intensity peaks were used for MS/MS analysis using LTQ Orbitrap velos Pro. Finally, MaxQuant software version 1.4.1.2 was used for data analysis as previously described29-30.
Western Blotting
The expression of proteins of interest was evaluated by Western blotting as we described previously31. Whole cell lysates were extracted and separated on SDS-PAGE, followed by immunoblotting against antibodies. Immunoreactive bands were visualized using Bio-Rad system (Bio-Rad, USA) with enhanced chemiluminescence (Bio-Rad).
Statistical Analysis
Data are presented as the mean ±SD and analyzed by Student's t-test for comparison of two groups, or by one-way analysis of variance (ANOVA) followed by Tukey's post-hoc test for multiple comparison. Statistical significance was considered at *p<0.05, **p<0.01, ***p<0.001.
Results
YSY01A suppresses MCF-7 cells survival
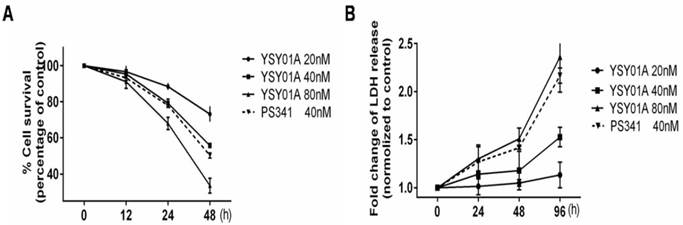
To determine whether YSY01A suppresses the survival of human breast cancer cells MCF-7, the cell viability was assessed by SRB assay following treatment with 20, 40 and 80 nM YSY01A for 12, 24 and 48 hrs (Fig. 1A). The compound inhibited survival of MCF-7 cells in concentration- and time‑ dependent manners with significant difference observed at 24 and 48 hrs. Compared to vehicle control (PBS), 20, 40 and 80 nM YSY01A were found to reduce cell survival by 11.6%, 20.8% and 32.0% at 24 hrs as well as 26.8%, 44.2% and 66.4% at 48 hrs, respectively (p< 0.001 except for 11.6%). To further detect the cytotoxicity of YSY01A, LDH (lactate dehydrogenase) release, indicative of rupture of cell membranes following treatment, was measured in MCF-7 cells exposed to 20, 40 and 80 nM YSY01A for 24, 48 and 96 hrs. The results revealed that treatment with 80 nM YSY01A for 96 hrs raised LDH release by 69.7% and became 2.31 fold with statistical significance as compared with vehicle control (p < 0.01). At 24 and 48 hrs, although the LDH release increased in a concentration- and time-dependent manner, significant difference was not observed between YSY01A and vehicle control (Fig. 1B), suggesting that YSY01A inhibited the survival of MCF-7 cells with limited cell damage, so we speculate that cell cycle progression may be susceptible for YSY01A treatment.
YSY01A inhibits survival of MCF-7 cells. MCF-7 cells were treated with either the vehicle (PBS), YSY01A 20, 40, 80 nM or PS341 40 nM for 12, 24 and 48 hrs (A) or 24, 48 and 96 hrs (B) followed by sulforhodamine B (SRB) assay (A) or CytoTox96® assay (B). Data are represented as the mean of three independent experiments performed in triplicate. ***p < 0.001
YSY01A induces cell cycle arrest on G2 phase in MCF-7 cells
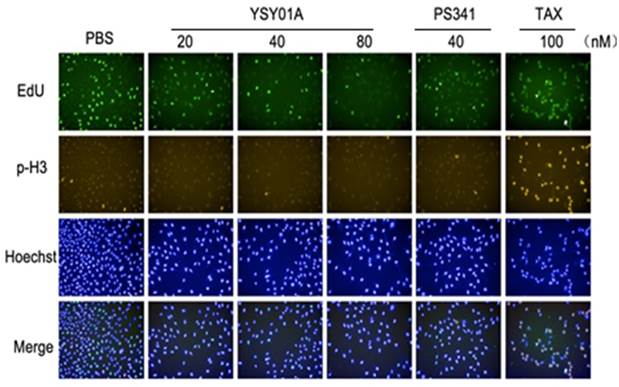
To investigate whether cell cycle arrest may contribute to loss of cell viability after YSY01A treatment, we further performed a cell cycle analysis using HCS. As shown in Fig. 2, increasing concentrations of YSY01A resulted in a dose-dependent rise in the relative NINI (normalized integrated nuclear intensity) value that corresponds to G2 phase. Exposure to YSY01A for 24 hrs caused an accumulation of cells on G2 phase from 28.6% (PBS) to 36.0% (40 nM) and 41.8% (80 nM) (Table 1). Taxol (Tax), which stabilizes microtubules by reducing their dynamicity and promotes mitotic arrest, was used as a positive control. Exposure to 100 nM Taxol caused M phase arrest in MCF-7 cells as determined by an increase in the NINI values. Thus, cell cycle arrest on G2 phase probably contributes to suppression of cancer cell survival in MCF-7 cells.
YSY01A induces cell cycle arrest in MCF-7 cells as determined by high-content imaging. Representative images of MCF-7 cells stained with EdU (5-ethynyl-2'-deoxyuridine)(a marker of S phase), p-H3 (a marker of M phase) and Hoechst 33342 (a marker of DNA), and the merge image also were shown. TAX represented the microtubules stabilizer taxol.
Percentage induction cell cycle arrest in MCF-7
| Group | Concentration (nM) | % Percentage of cell cycle distribution (mean±SD) | |||
|---|---|---|---|---|---|
| G1 | S | G2 | M | ||
| PBS | 0 | 26.8±4.7 | 43.3±3.8 | 28.6±1.9 | 1.5±0.6 |
| YSY01A | 20 | 17.2±1.3 | 53.9±1.8 | 28.3±0.8 | 0.5±0.2 |
| 40 | 34.1±1.6 | 28.1±3.9 | 36.0±4.5 | 1.7±0.8 | |
| 80 | 18.6±0.2 | 38.8±1.2 | 41.8±1.4** | 0.8±0.0 | |
| PS341 | 40 | 21.9±5.5 | 46.9±0.5 | 30.9±4.8 | 0.3±0.2 |
| TAX | 100 | 22.9±3.9 | 32.4±4.3 | 28.9±5.1 | 15.6±6.2* |
YSY01A induces cell cycle arrest in MCF-7 cells as determined by high-content imaging. Cells were treated in triplicate at noted concentrations of YSY01A for 24 h. Distribution percentage of cell cycle phases based on NINI (normalized integrated nuclear intensity values) derived from the DNA binding dye (Hoechst 33342). NINI value is calculated by measuring the integrated nuclear intensity divided by the number of nuclei within each well. The NINI derived from mitotic nuclei (p-H3 labeled) were indicative of 4N DNA content, while NINI values for S phase (EdU labeled nuclei) were ranged between 0.5 and 1X NINI values of p-H3 (2N to 4N DNA content), and images were analyzed using Columbus software. *p< 0.05, **p< 0.01.
YSY01A affects PI3K pathway and cell cycle associated proteins
PI3K and cell cycle associated proteins were changed significantly by YSY01A in LFQP assay
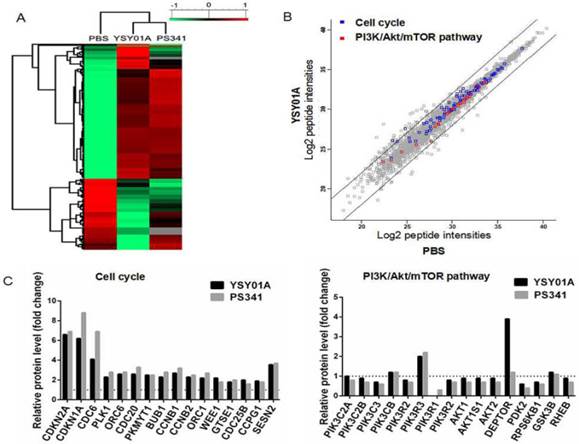
To extensively explore molecular mechanisms of YSY01A, LFQP offers a robust means for obtaining proteome profiles of YSY01A (Fig. 3A). The results showed that YSY01A was involved in regulation of apoptosis pathway, p53 associated pathway, mitochondrial dysfunction, endoplasmic reticulum stress, ubiquitin pathway, etc. PI3K pathway and cell cycle associated proteins were two of the most valuable and dominant fraction affected. The information of all proteins in the presence of 40 nM dose of YSY01A and PS341 were identified (Additional file 2: Supplemental data), and 395 peptides of 7560 whose abundances changed by more than two folds were identified (Fig. 3A&B). As shown in Fig. 3C, CDKN2A (p16), CDKN1A (p21Cip1) became 6.6 and 6.2 fold compared to vehicle control following exposure to YSY01A, while G2/M phase markers, CCNB1 (Cyclin B1) and CCNB2 (Cyclin B2), showed a 2.7 and 2.3 fold increase. Kinases in G2/M phase including CDC-6, CDC-20 and BUB1 exhibited a rise of 4.1, 2.6 and 2.3 fold, respectively, and YSY01A also increased levels of Wee1 to 2.2 fold. Sestrin 2 (SESN2), provides an important link between genotoxic stress, p53 and the mTOR signaling pathway, showed a 3.6 fold increase. And the profiles of PS341 were roughly coincident with YSY01A, apart from more intense enhancement in p16, p21Cip1 (6.9 and 8.8 fold change) and more mild increase in Wee1 (1.8 fold) compared with YSY01A. Moreover, quantitative analysis indicated that YSY01A may regulate PI3K pathway. For example, PIK3C3, Akt1 and PDK2 declined by 25.6%, 14.8%, 38.7% and p70S6K (RPS6KB1) decreased by 29.2%, whereas Deptor, an upstream inhibitor of mTORC1, was increased by 3.9 fold. The profiles of PS341 were also consistent with YSY01A except a slight increase in Deptor by 1.20 fold. These results were indicative of the potential involvement of YSY01A in regulation of cell cycle progression and PI3K pathway.
YSY01A changes PI3K and cell cycle associated proteins in MCF-7 cells revealed by LFQP (label-free quantitative proteomics). (A) Symmetrical matrix/non-clustering heatmap of Pearson correlation coefficients values from the significantly changed peptide intensity. Of 7560 peptides (image not shown in A), 395 showed abundance change of more than two folds. Normalized protein abundance values (Z-scores) were indicated colorimetrically for each protein in each sample ranging from green (low correlation, minimum, -1 ), to black(median, 0), and red (high correlation, maximum 1) (B) Scatter plot of all the log2 peptide intensities plotted against PBS group. The two oblique line showed Y=X+2 and Y=X-2, separating proteins at a ratio of 2 or 0.5 compared to PBS. The blue spots and red spots represented peptides of cell cycle proteins and PI3K/Akt/mTOR pathway. (C) Histograms of relative peptides quantitative levels of cell cycle and PI3K/Akt/mTOR pathway. The value of PBS was defined as 1 and indicated by a dash line. Sets of LC-MS/MS data were collected in triplicate following YSY01A treatment of 24 hrs.
YSY01A regulates expression of cell cycle, PI3K/Akt associated proteins and ERα
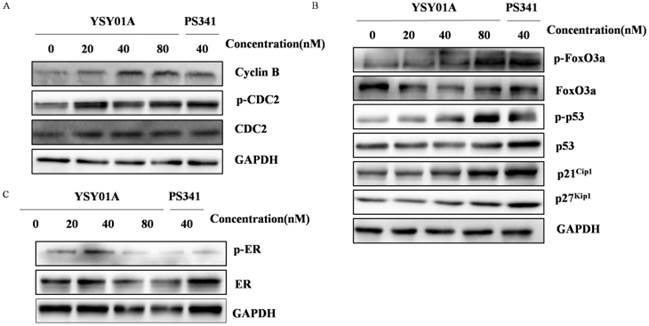
To validate molecular profiles of YSY01A identified by LFPQ, the G2/M phase marker, Cyclin B, CDC-2 and p-CDC-2 were firstly assessed by Western blotting. As shown in Fig. 4A, Cyclin B, CDC-2 and p-CDC-2 were all increased in MCF-7 cells treated with YSY01A, PS341 showed a similar trend as YSY01A with a slight increase in Cyclin B, and the similar results were obtained following YSY01A treatment at different time (Additional file 1: Fig. S1A). For the pivotal proteins involved in cell cycle transition, p53 were increased following YSY01A treatment, and the expression of p21Cip1 and p27Kip1 increased in the presence of 40 nM and 80 nM YSY01A (Fig. 4B). Moreover, it was not difficult to find that the bands began increasing at 3 hrs and reached a peak at 24 hrs (Additional file 1: Fig. S1B). However, the level of FoxO3a was decreased while its phosphorylation level was increased significantly. There was no obvious difference between PS341 and YSY01A, except PS341 treatment showed a more dramatic alternation in p53 and p27Kip1 than YSY01A. The increase of p21Cip1, p27Kip1 and p53 expression and activity caused by YSY01A suggested that the molecular change was significant for cell cycle arrest following YSY01A treatment.
YSY01A induces pivotal proteins involved in cell cycle transition and ERα in MCF-7 cells. After treatment of 20, 40, 80 nM YSY01A for 24 hrs or 40 nM PS341 for 24 hrs, expression of the cell cycle markers (A), cell cycle proteins (B) and ERα (C) were evaluated. Cell lysates were fractionated on SDS-polyacrylamide gels and analyzed by Western blotting with antibodies against cell cycle associated proteins. GAPDH was used as a loading control.
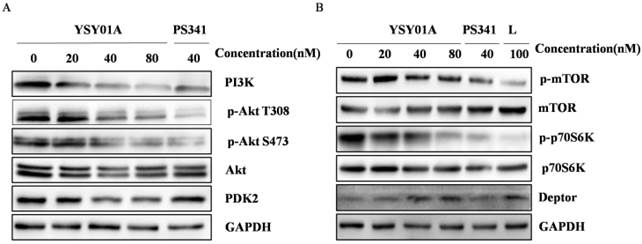
Considering the potential role of YSY01A in regulation of PI3K pathway, the phosphorylation of Akt, an activator of mTOR, and its downstream effector p70S6K were also evaluated by Western blotting in MCF-7 cells following YSY01A treatment. As shown in Fig. 5A, 40 and 80 nM YSY01A significantly inhibited the phosphorylation of Akt (Ser473 and Thr308), and the inhibition appeared starting a treatment of 3 hrs (Additional file 1: Fig. S2A). Furthermore, Deptor, a depressor of mTOR, was definitely raised after exposure to 40 and 80 nM YSY01A for 24 hrs, and 40 nM PS341 induced the same outcome as well (Fig. 5B & S2C). Since Akt promotes cell cycle progression, we speculate that the decreased phosphorylation level of Akt may result in cell cycle arrest in MCF-7 cells treated withYSY01A or PS341.
YSY01A modulates expression of PI3K pathway-related proteins. MCF-7 cells were treated with YSY01A at 20, 40, 80 nM for 24 hrs as directed and then subject for Western blotting analysis. L represented the positive control medicine rapamycin. GAPDH was used as a loading control.
Additionally, Western blotting revealed that ERα and p-ERα was down-regulated by YSY01A and PS341 (Fig. 4C & S1C). ERα is frequently overexpressed in the early stage of breast cancer and committed to modulate cell cycle progression, suggesting the response of ER-positive cells to YSY01A.
Discussion
The UPS plays a vital role in many cellular functions by selectively degrading key regulatory proteins. By blocking the action of proteasomes, proteasome inhibitors modulates cellular complexes that break down proteins and have been studies in anti-tumor treatment. Pre-clinical studies have revealed that bortezomib can sensitize the responses of cancer cells to chemotherapy agents and/or radiation therapy, for example 5-fluorouracil, cisplatin32, however, its toxicity prevents the achievement of higher drug concentrations and thus limits the efficacy of bortezomib monotherapy in certain cancers. Carfilzomib is a more selective proteasome inhibitor that is structurally distinct from bortezomib with reduced peripheral neuropathy. United States Food and Drug Administration granted authorization to carfilzomib in 2012 for treatment of multiple myeloma33. Pre-clinical data also support future clinical evaluation of carfilzomib in B-cell lymphoma34. However, the present proteasome inhibitors are usually ineffective to most solid tumors, including breast cancer. The new proteasome inhibitor, YSY01A, represents our attempt to develop more potent and less toxic proteasome inhibitors, and available for more wide variety of tumors including solid tumor. Previous studies have demonstrated little toxicity of YSY01A in the tumor-bearing nude mice (data to be published). In this study, we found that YSY01A inhibits cell vitality and induces cell cycle arrest in human breast cancer cell line MCF-7, suggesting its potential use in antitumor therapy.
With the aid of LFQP, the molecular profiles of YSY01A was revealed as well. LFQP assay allows analysis of abundant data about the changes of proteins levels following YSY01A treatment via a robust and easy-to-implement label-free relative quantification approach. This technique also obviate the requirements of protein staining or peptide labeling35. We identified a total of 7,561 non-redundant proteins from the 15 highest peaks in MS/MS, indicative of 52 categories changed, including the ubiquitination process, protein degradation (proteasome), mTOR, ErbB, cell cycle, and apoptosis. However, this assay could cause an omission of low-abundance proteins that may play an important role in cells' life. In relative to the overall blueprint of intracellular proteins, Western blotting was used to validate the results of LFQP and make an excellent complement of the alteration of some low-level proteins.
Additionally, the combination therapy involving cyclophosphamide, doxorubicin, and 5-fluorouracil (5-FU) (CDF) is the most common chemotherapeutic regimen of breast cancer36. Nevertheless, the clinical usefulness of CDF is limited by its remarkably low therapeutic window and frequent eruption of drug resistance. Therefore, development of chemo-preventive and therapeutic agents is needed to provide more therapeutic options, especially specific to drug resistance caused by PI3K/Akt over-expression. It has been demonstrated that blockade of the PI3K/Akt pathway can produce profound alternations in downstream gene expression37, including p53, FoxO, p21, p27. It is worth to note that some reports are demonstrated that PS341 decreases drug resistance via altering DNA repair pathways38 and histone deacetylase39-40 in relapsed/refractory multiple myeloma. Moreover, a phase II study of the combination of endocrine treatment and bortezomib in patients revealed that resistance to anti-hormonal therapies may be the result of an activated NF-κB signalling pathway in breast cancer41. NF-κB signalling pathway was not significantly altered after YSY01A and bortezomib treatment in our LFQP assay and the previous study of our group[data to be published]. The main reason may be associated with the status of NF-κB, as NF-κB was enhanced when the cancer cells obtain the drug resistance42-43, and the next plan in our group will focus on the effect of YSY01A on drug resistance when combined with other drugs.
ERα is another important therapeutic target in ERα-positive cancer cells, and a number of studies have reported the ERα alteration with bortezomib treatment. In addition to the previously mentioned studies,3-8 bortezomib has been also found to cause a strong increase in p21Cip1 mRNA level because of inhibit the ERα expression in ERα-positive cells44. In our study, YSY01A significantly enhanced the expression of p21Cip1 and relatively slightly altered the status of ERα expression, although the activity of ERα (p-ERα) was significantly inhibited by YSY01A and PS341. For the reason, the positive feedback between ERα and p70S6K point to the potential cause, indeed, we observed that ERα and p70S6K activity were declined in MCF-7 cells following YSY01A and PS341 treatment.
In conclusion, our study reinforces the potential benefit of YSY01A in cancer treatment, and provides extensive description of the mechanism of YSY01A on cell cycle arrest. We suggest that YSY01A may inhibit cell cycle progression and up-regulate the tumor suppressor genes p53, p27kip1 and p21Cip1 by inhibition of PI3K/Akt pathway and ERα activity in MCF-7 cells. Further studies directed at this strategy will provide more insights into the approach to interrupt proteasome for potential antineoplastic therapy.
Supplementary Material
Additional File 1Figures S1-S2.
Supplemental data.
Acknowledgements
Funding for this project was provided by the Ministry of National Science and Technology National Major Scientific and Technological Special Project for “Significant New Drugs Development” Program (No. 2009 ZX0930010) and the State Key Program of National Natural Science Foundation of China (No. 81172915).
Competing Interests
The authors have declared that no competing interest exists.
References
1. D'Arcy P, Linder S. Molecular pathways: translational potential of deubiquitinases as drug targets. Clin Cancer Res. 2014;20(15):3908-14
2. Liu YB, Gao X, Deeb D. et al. Ubiquitin-proteasomal degradation of antiapoptotic survivin facilitates induction of apoptosis in prostate cancercells by pristimerin. Int J Oncol. 2014;45(4):1735-41
3. Johnsen SA, Gungor C, Prenzel T. et al. Regulation of estrogen-dependent transcription by the LIM cofactors CLIM and RLIM in breast cancer. Cancer Res. 2009;69(1):128-136
4. Fan M, Nakshatri H, Nephew KP. Inhibiting proteasomal proteolysis sustains estrogen receptor-alpha activation. Mol Endocrinol. 2004;18(11):2603-15
5. Lonard DM, Nawaz Z, Smith CL, O'Malley BW. The 26S proteasome is required for estrogen receptor-alpha and coactivator turnover and for efficient estrogen receptor-alpha transactivation. Mol Cell. 2000;5:939-48
6. Reid G, Hubner MR, Metivier R. et al. Cyclic, proteasome-mediated turnover of unliganded and liganded ERα lpha on responsive promoters is an integral feature of estrogen signaling. Mol Cell. 2003;11(3):695-707
7. Powers GL, Ellison-Zelski SJ, Casa AJ. et al. Proteasome inhibition represses ERα gene expression in ER+ cells- a new link between proteasome activity and estrogen signaling in breast cancer. Oncogene. 2010;29(10):1509-1518
8. Zhang H, Sun L, Liang J. et al. The catalytic subunit of the proteasome is engaged in the entire process of estrogen receptor-regulated transcription. EMBO J. 2006;25:4223-33
9. Deroo BJ, Korach KS. Estrogen receptors and human disease. J Clin Invest. 2006;116(3):561-70
10. Ali S, Coombes RC. Endocrine-responsive breast cancer and strategies for combating resistance. Nat Rev Cancer. 2002;2(2):101-12
11. Josh Lauring, Ben Ho Park, Antonio C. Wolff. The Phosphoinositide-3-Kinase-Akt-mTOR Pathway as a Therapeutic Target in Breast Cancer. J Natl Compr Canc Netw. 2013;11(6):670-678
12. Sanchez CG, Ma CX, Crowder RJ. et al. Preclinical modeling of combined phosphatidylinositol-3-kinase inhibition with endocrine therapy for estrogen receptor-positive breast cancer. Breast Cancer Res. 2011;13(2):R21
13. Mahadevan D, Chiorean E.G, Harris WB. et al. Phase I pharmacokinetic and pharmacodynamic study of the pan-PI3K/mTORC vascular targeted pro-drug SF1126 in patients with advanced solid tumours and B-cell malignancies. Eur. J. Cancer. 2012;48(18):3319-3327
14. Liu CY, Shiau CW, Kuo HY. et al. Cancerous inhibitor of protein phosphatase 2A determines bortezomibinduced apoptosis in leukemia cells. Haematol. 2013;98(5):729-738
15. Chen KF, Yeh PY, Yeh KH. et al. Down-regulation of phospho-Akt is a major molecular determinant of bortezomib-induced apoptosis in hepatocellular carcinoma cells. Cancer Res. 2008;68(16):6698-6707
16. Kane RC, Dagher R, Farrell A. et al. Bortezomib for the treatment of mantle cell lymphoma. Clin Cancer Res. 2007;13(18 Pt 1):5291-5294
17. Morra E. Carfilzomib: a new opportunity for WM patients.Blood. Blood. 2014;124(4):468-9
18. Mohty M, Brissot E, Savani BN, Gaugler B. Effects of bortezomib on the immune system: a focus on immune regulation. Biol Blood Marrow Transplant. 2013;19(10):1416-20
19. Hideshima T, Richardson P, Chauhan D. et al. The proteasome inhibitor PS-341 inhibits growth, induces apoptosis, and overcomes drug resistance in human multiple myeloma cells. Cancer Res. 2001;61(7):3071-3076
20. Julian Adams. THE PROTEASOME: A SUITABLE ANTINEOPLASTIC TARGET. Nat Rev Cancer. 2004;4(5):349-60
21. Nooka AK. Management of Hematologic Adverse Events in Patients With Relapsed and/or Refractory Multiple Myeloma Treated With Single-Agent Carfilzomib. Oncology (Williston Park). 2013;1:27
22. Sandra E. Kurtin, Elizabeth Bilotti. Novel Agents for the Treatment of Multiple Myeloma: Proteasome Inhibitors and Immunomodulatory Agents. J Adv Pract. 2013;4(5):307-321
23. Engel RH, Brown JA, Von Roenn JH. et al. A phase II study of single agent bortezomib in patients with metastatic breast cancer: a single institution experience. Cancer Invest. 2007;25(8):733-737
24. Dees EC, Orlowski RZ. Targeting the ubiquitin-proteasome pathway in breast cancer therapy. Future Oncol. 2006;2(1):121-135
25. Xu-Hua Wang, Bo Xu, Jing-Tao Liu, Jing-Rong Cui. Effect of β-escin sodium on endothelial cells proliferation, migration and apoptosis. Vascular Pharmacology. 2008;49:158-165
26. Ma W, Chen M, Kaushal S. et al. PLGA nanoparticle-mediated delivery of tumor antigenic peptides elicits effective immune responses. International Journal of Nanomedicine. 2012;7:1475-1487
27. Kota KP1, Benko JG, Mudhasani R. et al. High Content Image Based Analysis Identifies Cell Cycle Inhibitors as Regulators of Ebola Virus Infection. Viruses. 2012;4(10):1865-1877
28. Tarawneh R, Lee JM, Ladenson JH. et al. CSF VILIP-1 predicts rates of cognitive decline in early Alzheimer disease. Neurology. 2012;78(10):709-719
29. Zimmer JS, Monroe ME, Qian WJ, Smith RD. Advances in proteomics data analysis and display using an accurate mass and time tag approach. Mass Spectrom Reviews. 2006;25(3):450-486
30. Yan Fu, Li-Yun Xiu, Wei Ji. et al. AMT: A Statistical Algorithm for Fast Detection of Protein Modifications From LC-MS/MS Data. Technological Innovation and Resources. 2011
31. Hou L, Xu B, Guo W. et al. Pseudolaric acid B inhibits inducible cyclooxygenase-2 expression via downregulation of the NF-κB pathway in HT-29 cells. J Cancer Res Clin Oncol. 2012
32. Boccadoro M, Morgan G. Cavenagh, J. Preclinical evaluation of the proteasome inhibitor bortezomib in cancer therapy. Cancer Cell International. 2005;5(1):18
33. Andreu-Vieyra C, Berenson JR. Carfilzomib in multiple myeloma. Expert Opin Biol Ther. 2014;29:1-15
34. Gu JJ, Hernandez-Ilizaliturri FJ, Kaufman GP. et al. The novel proteasome inhibitor carfilzomib induces cell cycle arrest, apoptosis and potentiates the anti-tumour activity of chemotherapy in rituximab-resistant lymphoma. British Journal of Haematology. 2013;162(5):657-669
35. Elliott MH, Smith DS, Parker CE, Borchers C. Current trends in quantitative proteomics. J Mass Spectrom. 2009;44(12):1637-1660
36. Ibrahim AS, Sobh MA, Eid HM. et al. Gingerol-derivatives: emerging new therapy against human drug-resistant MCF-7. Tumour Biology. 2014;4(2):39-47
37. Engelman JA1, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nature Reviews Genetics. 2006;7(8):606-19
38. Claire Gourzones-Dmitriev, alboukadel Kassambara, Surinder Sahota. et al. DNA repair pathways in human multiple myeloma. Cell cycle. 2013;12(17):2760-2773
39. Roman Hajek, David Siegel, Robert Z. Orlowski, et al. The role of histone deacetylase inhibitors in patients with relapsed/refractory multiple myeloma. Leukemia & Lymphoma. 2014;55(1):11-18
40. Iida S, Ri M. Determinants of sensitivity to proteasome inhibitors and strategies to overcome acquired resistance tobortezomib in multiple myeloma. Rinsho Ketsueki. 2014Mar;55(3):304-10
41. Trinh XB, Sas L, Van Laere SJ. et al. A phase II study of the combination of endocrine treatment and bortezomib in patients with endocrine-resistant metastatic breast cancer. Oncology Reports. 2012;27(3):657-63
42. Erstad DJ, Cusack JC Jr. Targeting the NF-κB pathway in cancer therapy. Surg Oncol Clin N Am. 2013;22(4):705-46
43. Li X, Abdel-Mageed AB, Mondal D, Kandil E. The nuclear factor kappa-B signaling pathway as a therapeutic target against thyroid cancers. Thyroid. 2013;23(2):209-18
44. Maynadier M, Shi J, Vaillant O. et al. Roles of estrogen receptor and p21(Waf1) in bortezomib-induced growth inhibition in human breast cancer cells. Molecular Mol Cancer Res. 2012;10(11):1473-81
Author contact
![]() Corresponding authors: Jingrong Cui, Email: jrcuiedu; Runtao Li, Email: lirtedu.cn.
Corresponding authors: Jingrong Cui, Email: jrcuiedu; Runtao Li, Email: lirtedu.cn.