Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2015; 6(3):270-275. doi:10.7150/jca.10638 This issue Cite
Research Paper
Amplification of Chromosome 8 Genes in Lung Cancer
Onur Baykara1, Burak Bakir1, Nur Buyru1, Kamil Kaynak2, Nejat Dalay3 ![]()
1. Department of Medical Biology, Cerrahpasa Medical Faculty, Istanbul University, Turkey
2. Department of Chest Surgery, Cerrahpasa Medical Faculty, Istanbul University, Turkey
3. Department of Basic Oncology, I.U. Oncology Institute, Istanbul University, Turkey
Received 2014-9-25; Accepted 2014-12-18; Published 2015-1-20
Abstract
Chromosomal alterations are frequent events in lung carcinogenesis and usually display regions of focal amplification containing several overexpressed oncogenes. Although gains and losses of chromosomal loci have been reported copy number changes of the individual genes have not been analyzed in lung cancer. In this study 22 genes were analyzed by MLPA in tumors and matched normal tissue samples from 82 patients with non-small cell lung cancer. Gene amplifications were observed in 84% of the samples. Chromosome 8 was found to harbor the most frequent copy number alterations. The most frequently amplified genes were ZNF703, PRDM14 and MYC on chromosome 8 and the BIRC5 gene on chromosome 17. The frequency of deletions were much lower and the most frequently deleted gene was ADAM9. Amplification of the ZNF703, PRDM14 and MYC genes were highly correlated suggesting that the genes displaying high copy number changes on chromosome 8 collaborate during lung carcinogenesis.
Keywords: Oncogenes, copy number changes, lung cancer
INTRODUCTION
Lung cancer is a major cause of cancer deaths throughout the world and is classified into two categories. Small cell lung cancer (SCLC) constitutes 15-20% of the lung cancers while non-small cell lung cancer (NSCLC) accounts for the remaining group. NSCLC includes squamous cell carcinoma, adenocarcinoma, adenosquamous cell carcinoma and large cell carcinoma [1, 2]. The development and progression of lung carcinoma is a long process that involves multiple cancer-related genes, as well as environmental and host genetic factors. Advances in molecular biology have shown that lung cancer is caused by the complex interaction of genetic and environmental factors and by accumulation of multiple genetic alterations such as amplifications, deletions, mutations and epigenetic events [2-4]. Although smoking is the main cause of lung cancer approximately 10% of cases are never smokers [5]. Despite advances in the therapeutic modalities the overall 5-year survival rate is still less than 15% for NSCLC and even lower for SCLC [6].
Tyrosine kinase inhibitors (TKIs) are used commonly to treat patients harboring activating mutations in the tyrosine kinase domain of the epidermal growth factor receptor gene. Patients who respond to TKI treatment are usually those who have never smoked and have generally adenocarcinomas [6-8]. Furthermore, during inhibitor therapy these patients develop resistance to treatment [9-10]. Another group of patients are individuals carrying K-Ras mutations with a history of smoking. These patients are also associated with resistance to EGFR-TKIs [11, 12]. Therefore, patient survival has not improved significantly and many unanswered questions about the optimal use of TKIs still remain.
To achieve a better and more stable response combined analysis of the relevant pathways which are involved in the mechanism of killing the tumor cells by chemotherapy is necessary. Identification of key molecular events during lung carcinogenesis and tumor progression will facilitate characterization of new therapeutic targets and development of new treatment strategies. Therefore, numerous studies aim to identify new molecular biomarkers which may predict a better clinical outcome for the patients.
Like in other solid tumors, chromosomal aberrations represent critical molecular events in the pathogenesis of lung cancer and recurrent genomic alterations have been reported in NSCLC [13]. The loci of recurrent aberrations frequently display regions of focal amplification in a wide range of tumors. Most of these regions harbor several oncogenes and the number of the amplified genes as well as the level of amplification may differ depending on the type of the tumor [13, 14]. Genes overexpressed within these amplicons are attractive targets for therapy. Frequently, two or more of these amplicons are present in a given tumor, suggesting that genes in the two amplified regions may collaborate in the development of the transformed phenotype [15, 16].
In contrast to other high throughput analysis methods, multiplex ligation-dependent probe amplification (MLPA) is an inexpensive and reliable method for the simultaneous analysis of a high number of different loci [17]. Although gains of partial or whole chromosomal arms on 1q, 3q, 5p, 8p and 11q have been reported in NSCLC copy number changes of the genes localized in these loci have not been analyzed in lung cancer [18-20]. In this study in order to understand the molecular background and to explore new targets for prevention and therapy, we investigated gene copy number changes in 22 genes in a group of 82 NSCLC tumors and matched non-cancerous tissue samples using MLPA.
MATERIALS AND METHODS
Tumor tissue and the corresponding normal lung tissue samples were obtained from 82 patients undergoing surgery at the Istanbul University Cerrahpasa Medical Faculty, Department of Chest Surgery. The patients taken into the study had not undergone any previous treatment and were admitted to the hospital for removal of the tumor as primary treatment. DNA was obtained by digestion of the tissues using a DNA isolation kit (Roche Diagnostics DNA Isolation Kit-Mannheim, Germany) according to the manufacturer's instructions.
Copy number variations of 22 genes were analyzed by MLPA in 82 patients with NSCLC. MLPA analysis was performed by using 100 ng of genomic DNA and the SALSA MLPA P078-C1 Probemix kit (MRC-Holland, Amsterdam, Netherlands) according to the manufacturer's instructions. The list of the genes and the probes used in the study are shown in Table 1. The samples were analyzed in duplicate using the ABI 3100 Genetic Analyzer (Applied Biosystems, Foster City, CA, USA). Raw data were normalized using the GeneMapper program (Applied Biosystems, Foster City, CA, USA). Relative peak areas of the amplification products were evaluated using the Coffalyzer MLPA analysis software (MRC-Holland, Amsterdam, the Netherlands) and compared with results obtained from the healthy tissue specimens to calculate gene copy numbers. For genes analyzed with multiple probes, the mean of all the peak values for this gene was calculated. A mean value below 0.75 was defined as loss, values between 0.75 and 1.25 were defined as normal, between 1.25 and 1.65 as heterozygote duplications, between 1.65 and 2.3 as triplications and between 0.25 and 0.75 as heterozygote deletions.
List of the genes analyzed by MLPA in the tumor and matched normal tissue samples.
| GENE | Chromosome | Description |
|---|---|---|
| ESR1 | 6q25 | Transcription factor |
| EGFR | 7p11 | Receptor tyrosine kinase in signaling |
| ZNF703 | 8p11 | Transcription factor in migration and proliferation |
| FGFR1 | 8p11 | Receptor tyrosine kinase in signaling |
| ADAM9 | 8p11 | Metalloproteinase in protein metabolism |
| IKBKB | 8p11 | Serine threonine kinase in signaling |
| PRDM14 | 8q13 | Transcription regulatory protein |
| MTDH | 8q22 | Transcription co-activator |
| MYC | 8q24 | Transcription factor in apoptosis and proliferation |
| CCND1 | 11q13 | Cell cycle control protein in signaling |
| C11ORF30 | 11q13 | Transcription regulatory protein |
| CDH1 | 16q22 | Adhesion molecule in signaling |
| CPD | 17q11 | Carboxylpeptidase in protein metabolism |
| MED1 | 17q12 | Component of mediator complex |
| ERBB2 | 17q12 | Receptor tyrosine kinase in signaling |
| CDC6 | 17q21 | Cell cycle control protein in signaling |
| TOP2A | 17q21 | DNA topoisomerase protein |
| MAPT | 17q21 | Microtubule assembly promoter |
| PPM1D | 17q23 | Serine threonine phosphatase |
| BIRC5 | 17q25 | Transcription factor in apoptosis and proliferation |
| CCNE1 | 19q12 | Cell cycle control protein in signaling |
| AURKA | 20q13 | Serine threonine kinase in signaling |
The study was approved by the Ethics Committee of Istanbul University, Cerrahpasa Medical Faculty and written informed consent was obtained from all patients enrolled in the study.
Statistical analysis was performed using the SPSS 21 for Windows (IBM Corp. Released version 2012. IBM SPSS Statistics for Windows, Version 21.0. Armonk, NY: IBM Corp.). We used the χ2 (2-tailed) test to analyze the association between gene copy number variations and clinicopathological characteristics and the Spearman's Rho test to evaluate the correlation between the most frequently amplified genes. p<0.05 was considered statistically significant.
RESULTS
To investigate whether there is a change in cancer-associated gene copy numbers in tumors from patients with non-small cell lung cancer, we performed MLPA analysis in 82 tumors and matched normal tissue samples. Amplification of genetic material was found in 69 of 82 (84.14%) of the NSCLC tissues analyzed by the MLPA assay, while the remainder of the tumors did not display amplification among the tested 22 genes (Table 2). Losses were observed in 21.9% of the patients.
CNV analysis of the 22 genes in NSCLC tumor tissue.
| Genes | No change | Duplication | Triplication | # of amplified samples | Heterozygote Deletion | Homozygote Deletion | Amplification % |
|---|---|---|---|---|---|---|---|
| ESR1 | 77 | 4 | 0 | 4 | 1 | 0 | 4.9 |
| EGFR | 67 | 7 | 8 | 15 | 0 | 0 | 18.3 |
| ZNF703 | 43 | 28 | 10 | 38 | 1 | 0 | 46.3 |
| FGFR1 | 68 | 6 | 3 | 9 | 5 | 0 | 11.1 |
| ADAM9 | 69 | 3 | 4 | 7 | 6 | 0 | 8.5 |
| IKBKB | 73 | 7 | 2 | 9 | 0 | 0 | 11.1 |
| PRDM14 | 58 | 24 | 10 | 34 | 0 | 0 | 41.5 |
| MTDH | 64 | 12 | 4 | 16 | 2 | 0 | 19.5 |
| MYC | 55 | 18 | 9 | 27 | 0 | 0 | 32.9 |
| CCND1 | 72 | 2 | 8 | 10 | 0 | 0 | 12.2 |
| C11ORF30 | 68 | 7 | 3 | 10 | 4 | 0 | 12.2 |
| CDH1 | 70 | 7 | 4 | 11 | 1 | 0 | 13.4 |
| CPD | 69 | 6 | 2 | 8 | 5 | 0 | 9.8 |
| MED1 | 62 | 9 | 5 | 14 | 4 | 2 | 17.1 |
| ERBB2 | 71 | 6 | 5 | 11 | 0 | 0 | 13.4 |
| CDC6 | 68 | 9 | 4 | 13 | 1 | 0 | 15.9 |
| TOP2A | 74 | 6 | 1 | 7 | 1 | 0 | 8.5 |
| MAPT | 75 | 3 | 2 | 5 | 2 | 0 | 6.1 |
| PPM1D | 62 | 12 | 4 | 16 | 4 | 0 | 19.5 |
| BIRC5 | 55 | 25 | 2 | 27 | 0 | 0 | 32.9 |
| CCNE1 | 64 | 13 | 5 | 18 | 0 | 0 | 22.1 |
| AURKA | 73 | 9 | 0 | 9 | 0 | 0 | 11.1 |
Chromosome 8 harbored the most frequent alterations. We used 7 and 5 probes hybridizing to the short and long arms of chromosome 8, respectively. Altogether, fifty five samples (67%) showed amplification of the ZNF703, PRDM14 and/or MYC genes. The most frequently amplified gene on the short arm of chromosome 8 was ZNF703 with a frequency of 46.3%. In 14 (17%) of these cases ZNF703 was the only amplified gene. PRDM14 and MYC genes on the long arm were amplified in 41.5% and 32.9% of the patients, respectively. Exclusive amplifications of the PRDM14 and MYC genes were present in 9 (10.9%) and 4 (4.9%) out of 55 cases, respectively. In the remaining 28 (34.1%) samples concurrent amplification of all or any two of these genes were observed at the same time (Table 3). 73.9%, 70.4% and 67% of these amplifications were duplications and 26.1%, 29.6% and 33.6% were triplications of the ZNF703, PRDM14 and MYC genes, respectively.
Frequency and coamplification of the most frequently amplified genes.
| GENES | n=82 (%) |
|---|---|
| ZNF703-PRDM14-MYC | 12 (14.63) |
| ZNF703-PRDM14 | 9 (10.97) |
| ZNF703-MYC | 4 (4.87) |
| PRDM14-MYC | 3 (3.65) |
| ZNF703 | 14 (17.07) |
| PRDM14 | 9 (10.97) |
| MYC | 4 (4.87) |
The frequency of gene amplifications in NSCLC tumors as determined by MLPA
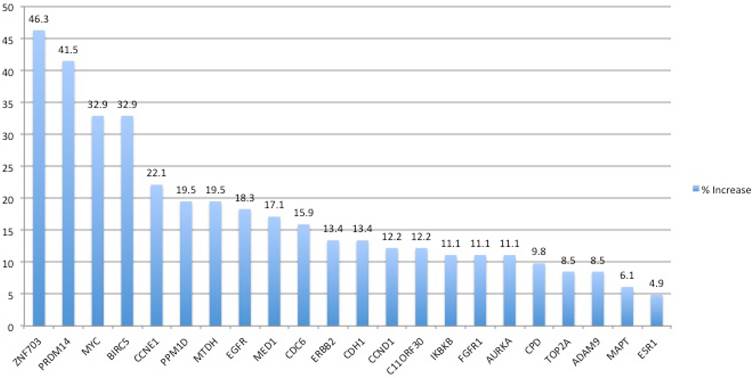
On the other hand, BIRC5 was the most frequently amplified gene on chromosome 17. There were 15 probes available hybridizing with 8 genes on the long arm of chromosome 17. Amplification of the BIRC5 gene was observed in 32.9% of the samples with the same frequency as for the MYC gene. The remaining genes analyzed in this study were amplified with frequencies varying between 4.9% and 22.1%. In 20 (24.3%) samples amplification of 7 or more genes were observed.
The mean copy numbers in the amplified samples were 1.31±0.36 for ZNF703, 1.27±0.36 for PRDM14, 1.29±0.46 for MYC and 1.23±0.25 for the BIRC5 genes. When we analyzed the correlation between the clinicopathological characteristics of the patients and gene amplification status, no association was observed with any clinical parameter (Table 4).
Distribution of CNVs in association with clinicopathological characteristics.
| CNV | p | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| n=82 | 1 gene | 2-3 genes | 4-6 genes | ≥ 7 genes | No change | Decreased | Total | ||
| Pathology | Adeno Ca | 5 (15.6) | 10 (31.3) | 5 (15.6) | 5 (15.6) | 6 (18.8) | 1 (3.1) | 32 | |
| SCC | 7 (20.0) | 7 (20.0) | 4 (11.4) | 11 (31.4) | 6 (17.1) | 0 (0) | 35 | 0.815 | |
| n/a | 4 (26.7) | 4 (26.7) | 2 (13.3) | 4 (26.7) | 1 (6.7) | 0 (0) | 15 | ||
| Stage | Stage 1A-2A | 3 (12.0) | 6 (24.0) | 3 (12.0) | 8 (32,0) | 5 (20.0) | 0 (0) | 25 | |
| Stage 2B-4 | 1 (6.3) | 2 (12.5) | 3 (18.8) | 6 (37.5) | 4 (25.0) | 0 (0) | 16 | 0.828 | |
| n/a | 12 (29.3) | 13 (31.7) | 5 (12.2) | 6 (14.6) | 4 (9.8) | 1 (2.4) | 41 | ||
| Gender | Male | 13 (18.6) | 16 (22.9) | 10 (14.3) | 17 (24.3) | 13 (18.6) | 1 (1.4) | 70 | |
| Female | 2 (22.2) | 4 (44.4) | 0 (0) | 3 (33.3) | 0 (0) | 0 (0) | 9 | 0.321 | |
| n/a | 1 (33.3) | 1 (33.3) | 1 (33.3) | 0 (0) | 0 (0) | 0 (0) | 3 | ||
| Age | < 50 years | 3 (30.0) | 2 (20.0) | 1 (10.0) | 3 (30.0) | 0 (0) | 1 (0) | 10 | |
| ≥ 50 years | 12 (17.6) | 17 (25.0) | 9 (13.2) | 17(25.0) | 13 (19.1) | 0 (0) | 68 | 0.560 | |
| n/a | 1 (25.0) | 2 (50.0) | 1 (25.0) | 0 (0) | 0 (0) | 0 (0) | 4 | ||
| Smoking Status | 30 packs/year | 6 (19.4) | 6 (19.4) | 4 (12.9) | 7 (22.6) | 8 (25.8) | 0 (0) | 31 | |
| 31-60 packs/year | 6 (21.4) | 6 (21.4) | 3 (10.7) | 9 (32.1) | 4 (14.3) | 0 (0) | 28 | ||
| >60 packs year | 1 (20.0) | 2 (40.0) | 1 (20.0) | 0 (0) | 1 (20.0) | 0(0) | 5 | 0.854 | |
| n/a | 3 (16.7) | 7 (38.9) | 3 (16.7) | 4 (22.2) | 0 (0) | 1 (5.6) | 18 | ||
The frequency of the deletions was considerably low (Table 2). The most frequently deleted gene was ADAM9 and was observed in 6 of 82 (7.3%) of the tumor samples while deletion of FGFR1 was observed in 5 (6.1%) tumors. The only gene showing a homozygous deletion was MED1 in 2 (2.4%) of the patients.
Finally, we applied multivariate analysis for the 4 most frequently altered genes to evaluate whether variations in these genes affect each other. A significant positive association was observed between the PRDM14 and ZNF703 (p=0.00), MYC (p=0.01) and BIRC5 (p=0.01) genes. A positive correlation was also present between the ZNF703 and MYC genes (p=0.04). These data indicate that copy number variations in the genes located on chromosome 8 may affect other genes located on the same chromosome.
DISCUSSION
Molecular cytogenetic methods such as fluorescence in situ hybridization (FISH), comparative genomic hybridization (CGH), and spectral karyotyping (SKY) have greatly improved our abilities to detect genome-wide copy number alterations in solid tumors [21, 22]. Genome-wide and massively parallel sequencing analyses have shown the presence of recurrent abnormalities in different regions including amplifications in a broad range of epithelial cancers [16]. Therefore, to develop more accurate diagnostic and therapeutic strategies, several investigators have focused on the identification of chromosomal aberrations associated with NSCLC using CGH or FISH [13, 14, 23, 24]. Results of these investigations have shown that recurrent genomic alterations such as gains of partial or whole chromosomal arms on several chromosomes along with losses of others are present in NSCLC. However, the resolution of conventional CGH is not sufficient for the precise identification of the submicroscopic molecular changes on the gene level [21]. In recent years a new method of high-throughput molecular analysis, the multiplex ligation-dependent probe amplification (MLPA) has been introduced. However, the potential utility of this approach has not been evaluated for the analysis of gene copy numbers in lung cancer yet. There is only a single report in the literature using MLPA for methylation analysis in lung tumors [25]. Since it requires only small quantities of DNA and is much more sensitive and reliable than cytogenetic analysis we performed MLPA analysis to investigate the copy number alterations in matched tumor tissues [17].
Among the 8 chromosomes which were investigated in this study chromosome 8 was the most frequently amplified chromosome. In previous studies numerical chromosomal aberrations have been reported for chromosome 8. However, the MYC oncogene has been reported as the most frequently amplified gene in various tumor types [26]. On the other hand, Kubokura et al. reported that chromosome 8 copy number alterations were not associated with MYC amplification in NSCLC [27]. In this study we analyzed 7 different genes on the long and short arms of chromosome 8. Although one of the amplified genes on the long arm of chromosome 8 was MYC it was not the primarily amplified gene on chromosome 8. The most frequently amplified genes were the ZNF703 (zinc finger protein 703) gene on the short arm, and PRDM14 on the long arm of chromosome 8.
ZNF703 is a member of the NET/NIZ family of transcription factors and has been identified recently as a novel oncogene in human breast cancer [16, 28, 29]. It has been characterized as the genetic driver of the A1 amplicon on chromosome 8 and has been implicated in different properties of the cancer cells including renewal, proliferation and invasion [30]. Overexpression of ZNF703 in lung cancer is in accordance with data observed in breast and gastric tumors [30-32]. Amplification of ZNF703 has been found to be second only to erbB2 and CCND1 genes in breast cancer and it has been shown that ZNF703 overexpression increases genome instability and contributes to tumor aggressiveness in breast cancer [16, 33]. More recently, enhanced ZNF703 expression has been correlated with repression of E-cadherin and increased lung metastasis rates in breast cancer [34]. These findings indicate that ZNF703 may function as an oncogene in different types of cancer. It remains to be determined how the expression and function of the ZNF703 protein is regulated.
The second most frequently amplified gene in our series was PRDM14 which is located on 8q13. PRDM14 is a member of the PRDM family of transcriptional regulators and controls pluripotency and epigenetic reprogramming by repressing/activating relevant genes through a variety of different mechanisms [35, 36]. PRDM14 is one of the key transcriptional regulators of primordial germ cell specification and over-expression of PRDM14 has been reported in different cancers [37-40]. Recently, the PRDM14 gene has been identified as a susceptibility locus for cancer [41]. Its amplification and over-expression has been associated with a more aggressive phenotype and reduced sensitivity to chemotherapy in breast cancer [37]. There are only two studies in the literature investigating the expression rate of PRDM14 in NSCLC tumors by immunohistochemistry and western blotting reporting that PRDM14 is strongly expressed in NSCLC [42, 43]. The authors also report a correlation between PRDM14 expression levels and differentiation. In our study group the frequency of tumors with high PRDM14 expression was lower than reported by Liu et al. [42] but in line with Zhang et al. [43] who observed expression in 35 % of the tumors.
The remaining frequently amplified genes were the BIRC5 and MYC oncogenes in accordance with previous reports. Data from a recent study suggests that BIRC5 may exert its effect by modulating Arf6 expression [44]. There are many reports in the literature indicating overexpression of these two genes in NSCLC and other cancers [45-49]. However, the more interesting finding is the frequent co-amplification of the genes which are located on the same chromosome.
Some of the genes within the amplicons are oncogenes that drive the amplicon while others are passengers. In our study group none of the amplified genes are located on the same amplicon. However, we observed significant correlations between the amplified genes, suggesting a collaborative role of different amplicons in the tumorigenesis of NSCLC. Our data indicate that amplification of the multiple amplicons on chromosome 8 are an important and significant contributor to NSCLC pathogenesis since collaboration between oncogenes within the different regions of the same chromosome are observed. Contribution of the regions on chromosome 8 may provide a selective advantage for the transformed phenotype. Further studies will be required to identify the associated genes and their mutual interactions in order to elucidate their role in lung carcinogenesis.
Acknowledgements
This study has been supported by the Scientific and Technical Research Council of Turkey, Project Nr. 114Z489.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Siegel R, Naishadham D, Jemal A. Cancer Statistics, 2013. CA Cancer J Clin. 2013;63:11-30
2. Herbest RS, Heymach JV, Lippman SM. Lung Cancer. N Engl J Med. 2008;359:1367-1380
3. Risch A, Plass C. Lung cancer epigenetics and genetics. Int J Cancer. 2008;123:1-7
4. Braithwaite KL, Rabbitts PH. Multistep evolution of lung cancer. Semin Cancer Biol. 1999;9:255-265
5. Hecht SS. Tobacco smoke carcinogens and lung cancer. J Natl Cancer Inst. 1999;91:1194-1210
6. Pao W, Miller V, Zakowski M. et al. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci USA. 2004;101:13306-13311
7. Paez JG, Jänne PA, Lee JC. et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304:1497-1500
8. Lynch TJ, Bell DW, Sordella R. et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350:2129-2139
9. Kobayashi S, Boggon TJ, Dayaram T. et al. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med. 2005;352:786-792
10. Pao W, Miller VA, Politi KA. et al. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLos Med. 2005;2:e73
11. Le Calvez F, Mukeria A, Hunt JD. et al. TP53 and KRAS mutation load and types in lung cancers in relation to tobacco smoke: distinct patterns in never, former, and current smokers. Cancer Res. 2005;65:5076-5083
12. Pao W, Wang TY, Riely GJ. et al. KRAS mutations and primary resistance of lung adenocarcinomas to gefitinib or erlotinib. PLoS Med. 2005;2:e17
13. Ding L, Getz G, Wheeler DA. et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature. 2008;455:1069-1075
14. Zhao X, Weir BA, LaFramboise T. et al. Homozygous deletions and chromosome amplifications in human lung carcinomas revealed by single nucleotide polymorphism array analysis. Cancer Res. 2005;65:5561-5570
15. Chin K, DeVries S, Fridlyand J. et al. Genomic and transcriptional aberrations linked to breast cancer pathophysiologies. Cancer Cell. 2006;10:529-541
16. Spellman P, Gray J. A new treasure in breast cancer gene hunt. Nat Med. 2011;17:422-423
17. Schouten JP, McElgunn CJ, Waaijer R. et al. Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic Acids Res. 2002;30:e57
18. Berrieman HK, Ashman JN, Cowen ME. et al. Chromosomal analysis of non-small-cell lung cancer by multicolor fluorescent in situ hybridization. Br J Cancer. 2004;90:900-5
19. Luk C, Tsao MS, Bayani J, Shepherd F, Squire JA. Molecular cytogenetic analysis of non-small cell lung carcinoma by spectral karyotyping and comparative genomic hybridization. Cancer Genet Cytogenet. 2001;125:87-99
20. Björkqvist AM, Husgafvel-Pursiainen K, Anttila S. et al. DNA gains in 3q occur frequently in squamous cell carcinoma of the lung, but not in adenocarcinoma. Genes Chromosomes Cancer. 1998;22:79-82
21. Kallioniemi A, Kallioniemi OP, Sudar D. et al. Comparative genomic hybridization for molecular cytogenetic analysis of solid tumors. Science. 1992;258:818-821
22. Okami K, Cairns P, Westra WH. et al. Detailed deletion mapping at chromosome 9p21 in non-small cell lung cancer by microsatellite analysis and fluorescence in situ hybridization. Int J Cancer. 1997;74:588-592
23. Weis BA, Woo MS, Getz G. et al. Characterizing the cancer genome in lung adenocarcinoma. Nature. 2007;450:893-898
24. Kim TM, Yim SH, Lee JS. et al. Genome-wide screening of genomic alterations and their clinicopathologic implications in non-small cell lung cancers. Clin Cancer Res. 2005;11:8235-8242
25. Castro M, Grau L, Puerta P. et al. Multiplexed methylation profiles of tumor suppressor genes and clinical outcome in lung cancer. J Transl Med. 2010;8:86
26. Mitelman database of chromosome aberrations and gene fusions in cancer; 2009. Mitelman F, Johansson B, Mertens F. http://cgap.nci.nih.gov/Chromosomes/Mitelman
27. Kubokura H, Koizumi K, Yamamoto M, Tanaka S. Chromosome 8 copy numbers and the c-myc gene amplification in non-small cell lung cancer. Analysis by interphase cytogenetics. Nihon Ika Daigaku Zasshi. 1999;66:107-112
28. Sircoulomb F, Nicolas N, Ferrari A. et al. ZNF703 gene amplification at 8p12 specifies luminal B breast cancer. EMBO Mol Med. 2011;3:153-166
29. Holland DG, Burleigh A, Git A. et al. ZNF703 is a common Luminal B breast cancer oncogene that differentially regulates luminal and basal progenitors in human mammary epithelium. EMBO Mol Med. 2011;3:167-180
30. Zhang X, Mu X, Huang O. et al. Luminal breast cancer cell lines overexpressing ZNF703 are resistant to tamoxifen through activation of Akt/mTOR signaling. PLoS One. 2013;8:e72053
31. Yang G, Ma F, Zhang M. et al. ZNF703 acts as an oncogene that promotes progression in gastric cancer. Oncol Rep. 2014;31:1877-1882
32. Ma F, Bi L, Yang G. et al. ZNF703 promotes tumor cell proliferation and invasion and predicts poor prognosis in patients with colorectal cancer. Oncol Rep. 2014;32:1071-1077
33. Reynisdottir I, Arason A, Einarsdottir BO. et al. High expression of ZNF703 independent of amplification indicates worse prognosis in patients with luminal B breast cancer. Cancer Med. 2013;2:437-446
34. Slorach EM, Chou J, Werb Z. Zeppo1 is a novel metastasis promoter that represses E-cadherin expression and regulates p120-catenin isoform expression and localization. Genes Dev. 2011;25:471-484
35. Hohenauer T, Moore AW. The PRDM family: expanding roles in stem cells and development. Development. 2012;139:2267-2282
36. Nakaki F, Saitou M. PRDM14: a unique regulator for pluripotency and epigenetic reprogramming. Trends Biochem Sci. 2014;39:289-298
37. Nishikawa N, Toyota M, Suzuki H. et al. Gene amplification and overexpression of PRDM14 in breast cancers. Cancer Res. 2007;67:9649-9657
38. Dettman EJ, Justice MJ. The zinc finger SET domain gene PRDM14 is overexpressed in lymphoblastic lymphomas with retroviral insertions at Evi32. PLoS One. 2008;3:e3823
39. Moelans CB, de Weger RA, Monsuur HN. et al. Molecular profiling of invasive breast cancer by multiplex ligation-dependent probe amplification-based copy number analysis of tumor suppressor and oncogenes. Mod Pathol. 2010;23:1029-1039
40. Simko SJ, Voicu H, Carofino BL. et al. Mouse lymphoblastic leukemias induced by aberrant Prdm14 expression demonstrate widespread copy number alterations also found in human ALL. Cancers. 2012;4:1050-1066
41. Ruark E, Seal S, McDonald H. et al. Identification of nine new susceptibility loci for testicular cancer, including variants near DAZL and PRDM14. Nat Genet. 2013;45:686-689
42. Liu B, Zhang S, Hui L. et al. Relationship between the expression of PRDM14 in non-small cell lung cancer and the clinicopathologic characteristics. Zhongguo Fei Ai Za Zhi. 2010;13:867-872
43. Zhang T, Meng L, Dong W. et al. High expression of PRDM14 correlates with cell differentiation and is a novel prognostic marker is resected non-small cell lung cancer. Med Oncol. 2013;30:605
44. Knizhnik AV, Kovaleva OB, Laktionov KK. et al. Arf6, RalA and BIRC5 protein expression in non small cell lung cancer. Mol Biol. (Mosk). 2011;45:307-315
45. Falleni M, Pellegrini C, Marchetti A. et al. Survivin gene expression in early stage non-small cell lung cancer. J Pathol. 2003;200:620-626
46. Zhang LQ, Wang J, Jiang F. et al. Prognostic value of survivin in patients with non-small cell lung carcinoma: a systematic review with meta-analysis. PLos One. 2012;7:e34100
47. Prochownik EV. c-Myc as a therapeutic target in cancer. Expert Rev Anticancer Ther. 2004;4:289-302
48. Yoo J, Kim CH, Song SH. et al. Expression of caspase-3 and c-myc in non-small cell lung cancer. Cancer Res Treat. 2004;36:303-307
49. Chen Z, Zheng H, Guo Y. et al. miRNA-145 inhibits non-small cell lung cancer cell proliferation by targeting c-Myc. J Exp Clin Cancer Res. 2010;29:151
Author contact
![]() Corresponding author: Prof. Dr. Nejat Dalay, I.U.Oncology Institute, 34093 Capa, Istanbul, Turkey. e-mail : ndalaycom; Phone : 90 542 2168861; Fax : 90 212 5348078
Corresponding author: Prof. Dr. Nejat Dalay, I.U.Oncology Institute, 34093 Capa, Istanbul, Turkey. e-mail : ndalaycom; Phone : 90 542 2168861; Fax : 90 212 5348078