Journal of Cancer
ISSN: 1837-9664
3.2
Impact Factor
ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2015; 6(2):184-191. doi:10.7150/jca.10822 This issue Cite
Research Paper
MiR-217 Promotes Tumor Proliferation in Breast Cancer via Targeting DACH1
Qiang Zhang1, Yonghui Yuan2, Jianchun Cui3, Tingting Xiao4, Daqing Jiang1 ![]()
1. Department of Breast Surgery, Liaoning Cancer Hospital & Institute, Shenyang, Liaoning Province, 110042, China.
2. Department of Infection, Liaoning Cancer Hospital & Institute, Shenyang, Liaoning Province, 110042, China.
3. Department of Endocrine Surgery, People's Hospital of Liaoning Province, Shenyang, Liaoning Province, 110042, China.
4. School of Chinese Medicine, Hong Kong Baptist University.
Received 2014-10-15; Accepted 2014-12-5; Published 2015-1-1
Citation:
Zhang Q, Yuan Y, Cui J, Xiao T, Jiang D. MiR-217 Promotes Tumor Proliferation in Breast Cancer via Targeting DACH1. J Cancer 2015; 6(2):184-191. doi:10.7150/jca.10822. https://www.jcancer.org/v06p0184.htm
Other stylesAbstract
Objective: The expression of DACH1 was frequently lost in human breast cancer, which significantly correlated with poor prognosis. Herein, we aim to investigate its underlying mechanisms.
Methods: The expression of miR-217 was detected by Taqman PCR. The mRNA and protein level of DACH1 were investigated by real time PCR and western blot. The dual-luciferase reporter system was used to determine the direct interaction between miR-217 and DACH1. A series of gain&loss of function assays were performed to measure the affects of miR-217 on tumor proliferation and cell cycle distribution.
Results: Compared to that in normal breast samples, the expression of miR-217 was significantly upregulated in breast cancer tissues. High level of miR-217 was notably correlated with highly histological grade, the triple negative subtype and advanced tumor stage. Moreover, the expression of miR-217 was negatively correlated with the expression of DACH1. The results of dual-luciferase reporter assay demonstrated that miR-217 directly targets and inhibits the transcriptive activity of DACH1. In vitro, treatment with miR-217 mimics significantly suppressed the proliferation of MCF-7 cells, induced G1 phase arrest and inhibited the expression of cyclin D1; while these effects were significantly reversed by the restoration of DACH1. In MDA-MB-231 cells, treatment with miR-217 inhibitors enhanced the cellular proliferation, promoted cell cycle progression and upregulated the expression of cyclin D1, which were neutralized by the pre-treatment of siRNA-DACH1. In vivo, inhibition of miR-217 significantly suppressed the xenografts growth and downregulated the expression of cyclin D1.
Conclusion: We found that miR-217 was commonly overexpressed in breast cancer, which could enhance tumor proliferation via promoting cell cycle progression. Moreover, the DACH1 (the cell fate determination factor) was identified as a novel target of miR-217. Our results proposed inhibiting miR-217 to be a potent therapeutic strategy for breast cancer.
Keywords: DACH1, miR-217, breast cancer, proliferation, cell cycle
Introduction
Globally, breast cancer have become the second leading cause of cancer-related deaths among the female population[1]. In 2014, there were estimated 232670 newly diagnosed and 40000 deaths caused by breast cancer in the United States[2]. In despite of that many advances have been made in breast cancer systemic treatment, a great amount of patients diagnosed with early stage breast cancer eventually presented tumor recurrence and metastasis and obtained a poor prognosis[3, 4]. Therefore, to unravel the molecular mechanisms underlying breast cancer initiation and progression was urgently required.
The DACH1 gene was found to be able to regulate normal development as a cell-fate determination factor[5, 6]. Up to date, dysregulation of DACH1 has been reported in various human malignancies including breast cancer, ovarian cancer, renal carcinoma and gastric cancer[7-10]. DACH1 is expressed in normal human breast epithelium, while reduced expression of DACH1 protein was found in a large cohort of human breast cancers, which was significantly correlated with poor prognosis[8]. Moreover, reintroduction of DACH1 significantly reversed the transformed phenotype in these human mammary epithelial cells transformed with oncogenes (such as c-Myc, ErbB2 and Ras)[11, 12]. These findings evidenced that DACH1 played an important role in breast cancer. In previous studies, DACH1 was found epigenetically silenced in colorectal cancer and clear cell renal cell carcinoma[9, 13]. However, the mechanism underlying frequent loss of DACH1 in breast cancer has not been explored yet.
MicroRNAs were a group of small non-coding RNAs which consisted of approximately 22 nucleotides[14]. According to previous studies, microRNAs were encoded within the genome and derived from these endogenous short hairpin precursors (processed by the Dicer and Drosha enzymes). The mature microRNAs were proved to able to negatively regulate the gene expression by either degrading target mRNAs or inhibiting their translation[15]. A large body of evidences suggested that microRNAs were involved in the manipulation of proliferation, differentiation, cell cycle, apoptosis, authophagy and metabolism[14-16].
In our recent study focusing on the different microRNA profile between normal and breast cancer, we detected that microRNA-217 (miR-217) was significantly overexpressed in breast cancer samples (unpublished yet). Interestingly, the bioinformatical analysis indicated there exista seed sequence of miR-217 at the 3'-UTR of the DACH1 gene. Based on these findings, we thus hypothesized that miR-217 might contribute to the development of breast cancer through directly targeting DACH1. In the present study, we demonstrated that miR-217 was progressively expressed in breast cancer and correlated with poor prognosis. More importantly, we found that miR-217 directly inhibited DACH1 and subsequently promoted cell cycle progression in breast cancer.
Materials and Methods
Samples collection
A panel of 39 breast cancer tissues (30 ductal carcinoma and 9 lobular carcinoma) and their adjacent normal tissues were collected from January 2008 to January 2009 at Liaoning Cancer Hospital (Shenyang, China). All the patients were pathologically diagnosed according with the WHO classification criteria. Our study was approved by the Ethical Committee of Liaoning Cancer Hospital and written consents were obtained from all patients. Patients' information was supplied in Table 1.
Table 1
Patients' characteristics.
| Variables | |
|---|---|
| Sample size | 39 |
| Age (years) | |
| Median | 49.2 |
| Range | 37-61 |
| Histology | |
| Ductal | 30 |
| Lobular | 9 |
| Subtype | |
| Triple negative | 9 |
| Non-triple negative | 30 |
| Histological grade | |
| I | 20 |
| II | 13 |
| III | 6 |
| Stage | |
| I | 15 |
| II | 12 |
| III | 8 |
| IV | 4 |
Cell line culture
MDA-MB-231, MCF-7, T47D and MDA-MB-468 were obtained from ATCC (American Type Culture Collection, MD, USA). The cells were routinely cultured in DMEM (Dulbecco's modified Eagle medium, Gibco, CA, USA) supplemented with 10% FBS (fetal bovine serum, Gibco), at 37°C with 5% CO2.
RNA extraction and qRT-PCR (quantitative reverse transcription polymerase chain reaction)
Total RNA was extracted using the Trizol reagent (Invitrogen, CA, USA). The expression of miR-217 was determined using Taqman microRNA assay kit (Applied Biosystems, CA, USA). U6B was set as the endogenous control. For quantification of DACH1, qRT-PCR was performed using SYBR Premix Ex Taw (Takara, Dalian, China). ß-actin was set as the endogenous control. The primers were: DACH1 forward: GGAATGGATTGTGGCTGAAC; DACH1 reverse: GGTATTGGACTGGTACATCAAG (122 bp). ß-actin forward: AGCACAATGAAGATCAAGATCAT; ß-actin reverse: ACTCGTCATACTCCTGCTTGC (127 bp). The relative mRNA expression was calculated by the 2-ΔΔCT method.
Western Blot
Cells were lysed by the RIPA buffer (Beyotime, Hangzhou, China) and the concentration was determined using a Pierce protein assay kit (Thermo Fisher, CA, USA). The total protein were separated on the SDS-PAGE, then the protein was transferred onto PVDF membranes (GE Health, UK) and blocked with 5% non-fat milk for two hours at room temperature. The membranes were incubated with primary antibodies overnight at 4°C. The specific protein bands were detected with secondary antibodies conjugated to horseradish peroxidase (1:3000, Cell Signaling Technology, MA, USA) and visualized using a chemiluminescent kit (Beyotime). The primary antibodies were anti‑cyclin D1 (1:100, Cell Signaling Technology) and anti-DACH1 (1:1000, Abcam, MA, USA). β-actin (1:2000, Cell Signaling Technology) was set as the internal control. The results were analyzed using the Quantity-One software (Bio-Rad, CA, USA).
Transfection
1×106 cells/well were plated into the six-well plates and incubated overnight. Then the cells were transfected with 100 nmol miR-217 mimics or miR-217 inhibitors using Lipofectamine 2000 (Invitrogen). Eight hours later, the medium was replaced by normal fresh medium. The miR-217 mimics and miR-217 inhibitors were all purchased from Genechem Biotechnology (Shanghai, China).
For small interfering RNA (siRNA)-mediated gene knockdown, 100 nmol DACH1 siRNA (5'-AAGGCCTCCTAAGAGGACTCA-3' and 5'-AAGGACTTCGAGACCCTCTAC-3') was transfected into MCF-7 cells using Lipofectamine 2000 according with the manufacturer's instructions. Twenty-four hours later, the miR-217 mimics were also transfected into MCF-7 cells, and these cells were collected 24 hours later for cell cycle analysis, 48 hours later for MTT assay and colony formation assay.
MTT Assay
5×103 cells/well were seeded into the 96-well plates and incubated overnight. Then the cells were transfected with miR-217 mimics or miR-217 inhibitors using Lipofectamine 2000. Forty-eight hours later, 5 µl MTT solution (5 mg/ml, Sigma-Aldrich, MO, USA) was added into each well and incubated for one hour and then was terminated by 200 µl DMSO. The absorption at 570 nm was measured on a microplate reader.
Colony formation assay
For the plate colony formation assay, 100 cells/well were seeded into the six-well plates and routinely cultured for two weeks. The cells were then fixed with 30% formaldehyde for 15 minutes and stained with 0.1% crystal violet. The colonies (containing more than 50 cells) number was determined under an optical microscope.
Cell cycle analysis
1×106 cells were fixed with 90% methanol for at least two hours and then resuspended in the buffer consisted of 0.1% BSA (bovine serum albumin, Sigma-Aldrich), 0.05% Triton X-100 (Sigma-Aldrich) and 50 μg/ml RNase A (Sigma-Aldrich). Then the cells were stained with PI solution (propidium iodide, 25 μg/ml, BD Bioscience, CA, USA) at dark for 15 minutes. Cell counting was performed on a FACS Calibur system (BD Biosciences) and the results were analyzed using Cell Quest Pro software (BD Biosciences).
Construction of the dual-luciferase reporter system
The wild-type and mutant 3'-UTR of DACH1 containing the seed sequence were synthesized and cloned into the pMIR-Report vector (Applied Biosystems). The wild-type 3'-UTR of DACH1 was mutated following the principle of complementary base pairing to disrupt the miR-217 binding sites. MCF-7 cells were transfected with miR-217 mimics (20 and 40 nmol) for 24 hours and then the cells were co-transfected with wild-type or mutated 3'-UTR of DACH1 using Lipofectamine 2000. Forty-eight hours later, cell lysates were incubated with dual-luciferase reagents (Promega, CA, USA) and the luciferase activities were determined by the luciferase assay kit (Promega).
The tumor xenografts growth assay
Our study was approved by the ethic committee for animal research at Liaoning Cancer Hospital. Six-eight weeks old female Balb/c nude mice were purchased from Slac Technology (Shanghai, China). In brief, 1×107 cells MDA-MB-231 cells with miR-217 inhibition (or negative control) were subcutaneously injected into to the posterior flank. Tumor diameters were measured once a week and tumor volume=(length×width2)/2. All the mice were sacrificed on the 28th day post-injection and the xenografts tissues were collected for immunohistochemical staining analysis.
Immunohistochemical staining assay
This assay was performed strictly as described in a previous publication[17]. In brief, the xenografts tissues were formalin-fixed/paraffin-embedded and cut into 4 μm slides. The antigen retrieval process was performed in a microwave for 25 minutes. The primary antibodies used were: anti‑Ki 67 (1:100, Cell Signaling Technology), anti‑cyclin D1 (1:200, Cell Signaling Technology) and anti-DACH1 (1:100, Abcam, MA, USA). Staining was performed using the immunohistostain IHC kit (Mingrui Biotechnology, Shanghai, China). The results of staining were evaluated by two pathologists separately.
Statistical analysis
All the in vitro experiments were performed in triplicate and repeated at least three times. The SPSS 17.0 (Chicago, IL, USA) software was used for statistical analysis. Data were expressed as mean±SD (standard deviation). Pair-wise comparisons were conducted by t-test. Serial analysis was performed using one-way ANOVA. P<0.05 was defined to be statistically significant.
Results
Expression of miR-217 was upregulated in breast cancer
Compared to the adjacent normal tissues, the expression of miR-217 was much higher in breast cancer samples (P<0.001, Figure 1A). High level of miR-217 was correlated with highly histological grade (P=0.018, Figure 1B), the triple negative subtype (P=0.015, Figure 1C) and advanced tumor stage (P=0.003, Figure 1D). All these findings indicated an important role of miR-217 in breast cancer.
MiR-217 directly targeted the 3'-UTR of DACH1
We then studied the correlation between miR-217 level and its candidate target---DACH1 in breast cancer tissues. Compared to those cases with low level of miR-217, the expression of DACH1 was much lower in the patients with high level of miR-217 (P<0.001, R2= 0.343, Figure 2A). Using the Targetscan software, we found a miR-217 binding site within the 3'-UTR of DACH1 (Figure 2B, upper part). Therefore, we then constructed the dual-luciferase reporter system to determine the direct interaction between miR-217 and 3'-UTR of DACH1 (Figure 2B, lower part). As our data shown, 20 nmol and 40 nmol miR-217 mimics suppressed the luciferase activities about 50% and 60% in the wild-type group (P=0.025 and 0.008, respectively, Figure 2C), while no obvious alteration was detected in the mutant group (Figure 2C), indicating that miR-217 could directly bind with DACH1 3'-UTR at the seed sequence.
MiR-217 promoted cellular proliferation and accelerated cell cycle progression via inhibiting DACH1 in MCF-7 cells.
As shown in Figure 3A, the expression of miR-217 was much higher in MDA-MB-231 and MDA-MB-468 cells than in MCF-7 and T47D cells. To investigate the biologic functions of miR-217, we transiently elevated (or downregulated) the expression of miR-217 in MCF-7 or MDA-MB-231 cells (P<0.001 and 0.011, respectively, Figure 3B). Compared to the negative control, miR-217 overexpression notably accelerated the cellular growth of MCF-7 cells (P=0.036, Figure 3C), which was partially reversed when DACH1 was pre-overexpressed (P=0.029, Figure 3C). Consistently, the colony formation was also enhanced by miR-217 overexpression (P=0.018, Figure 3D), which was significantly reversed by the DACH1 restoration (P=0.025, Figure 3D). Compared to the blank and negative control groups, miR-217 overexpression promoted more MCF-7 cells into S phase, accompanied with the decrease of G1 phase (P=0.013, Figure 3E), while pre-overexpression of DACH1 almost neutralized this phenomenon (P=0.045, Figure 3E). These alterations were accompanied with changes of DACH1 and cyclin D1 (Figure 3F).
Inhibition of miR-217 suppressed cellular growth and arrested cell cycle via upregulating DACH1 in MDA-MB-231 cells.
We then the investigated the affects of miR-217 inhibition on cellular proliferation and cell cycle distribution of MDA-MB-231 cells. Compared to the controls, miR-217 inhibition notably decelerated cellular growth in MDA-MB-231 cells (P=0.007, Figure 4A), which could be partially reversed when DACH1 was pre-inhibited (P=0.033, Figure 4A). In addition, the colony formation was also suppressed by miR-217 inhibition (P=0.036, Figure 4B), which was partially reversed by the DACH1 pre-inhibition (P=0.051, Figure 4B). Consistently, miR-217 inhibition induced G1 phase arrest and decreased the proportion of S phase in MDA-MB-231 cells (P=0.027, Figure 4C), which was significantly neutralized by the pre-inhibition of DACH1 (P=0.041, Figure 4C). The expression of cyclin D1 protein presented similar alterations (Figure 4D).
Inhibition of miR-217 suppressed tumor growth in vivo.
We further determined the affects of miR-217 inhibition on tumor growth in the nude mice model. As our data shown, treatment with miR-217 inhibitors significantly suppressed the growth of MDA-MB-231 xenografts (P=0.008, Figure 5B). Consistently, inhibition of miR-217 upregulated the level of DACH1 and downregulated the expression of Ki 67 and cyclin D1 (Figure 5B).
Figure 1
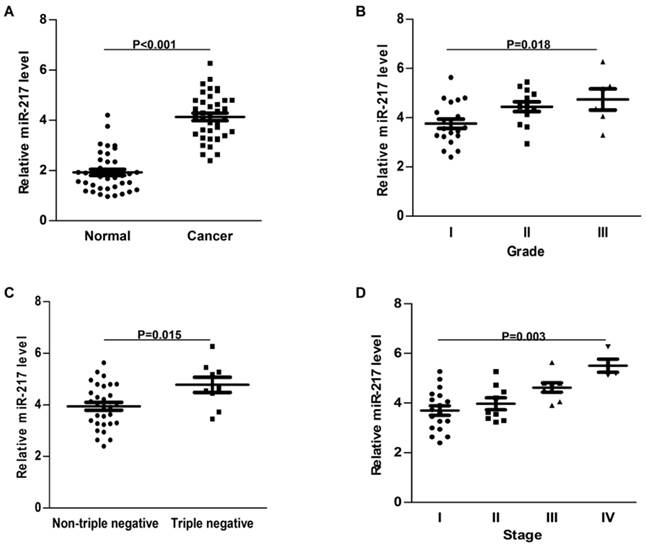
Expression of miR-217 was upregulated in breast cancer. (A) compared to the normal control, the expression of miR-217 was much higher in the breast cancer samples (P<0.001); (B) high level of miR-217 was positively correlated with histological grade (P=0.018); (C) the expression of miR-217 was much higher in triple negative breast cancer tissues (P=0.015); (D) high level of miR-217 was significantly correlated with advanced tumor stage (P=0.003).
Figure 2
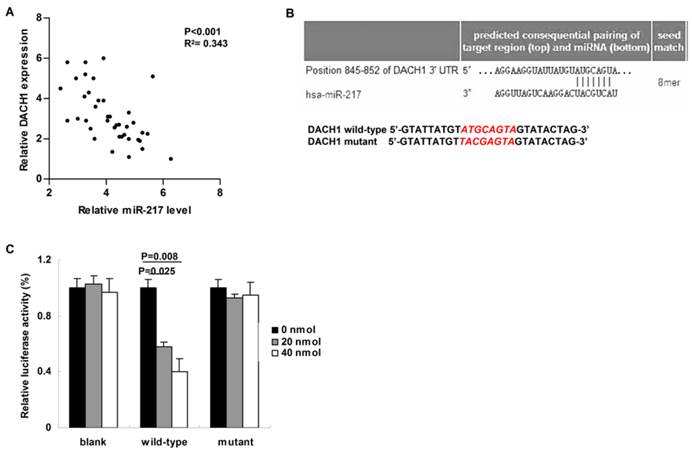
MiR-217 directly targeted and inhibited DACH1. (A) Compared to those cases with low level of miR-217, the expression of DACH1 was much lower in the patients with high level of miR-217 (P<0.001, R2=0.343); (B) the Targetscan software predicted a miR-217 binding site within the 3'-UTR of DACH1 (upper part). Then a wild-type and a mutant 3'-UTR of DACH1 were constructed to determine the interaction between miR-217 and 3'-UTR of DACH1 (lower part); (C) in MCF7 cells, 20 nM and 40 nM miR-217 mimics suppressed the luciferase activities about 50% and 60% in the wild-type group (P=0.025 and 0.008, respectively). No obvious alteration was detected in the mutant group.
Figure 3
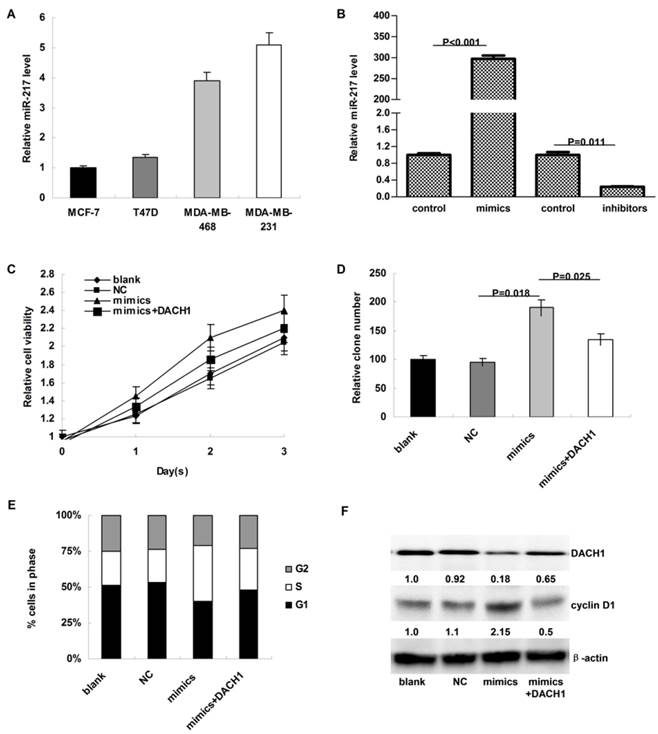
MiR-217 promoted cellular proliferation and accelerated cell cycle progression via inhibiting DACH1 in MCF-7 cells. (A) the expression of miR-217 was much higher in MDA-MB-231 and MDA-MB-468 cells than in MCF-7 and T47D cells (about 4-folds); (B) the transient transfection with specific mimics (or inhibitors) significantly upregulated (or downregulated) the expression of miR-217 (P<0.001 and 0.011, respectively); (C) compared to the negative control, miR-217 overexpression notably accelerated the cellular growth of MCF-7 cells (P=0.036), which was partially reversed when DACH1 was pre-overexpressed (P=0.029); (D) the colony formation were also enhanced by miR-217 overexpression (P=0.018), which was significantly reversed by the DACH1 restoration (P=0.025); (E and F) compared to the blank and negative control groups, miR-217 overexpression promoted more MCF-7 cells into S phase, accompanied with notably decrease of G1 phase (P=0.013), while pre-overexpression of DACH1 almost neutralized this phenomenon (P=0.045). These alterations were accompanied with changes of cyclin D1.
Discussion
Previous study demonstrated that DACH1 was significantly silenced in a panel of about 2,000 human breast cancer patients, and loss of DACH1 was dramatically reduced in invasive cancer compared to the non-invasive cases. Moreover, loss of DACH1 was significantly correlated with deaths from breast cancer 40 months earlier, indicating a key role of DACH1 in breast cancer[8]. Unfortunately, the underlying mechanisms have not been revealed yet. In the present study, we demonstrated that DACH1 was a direct target of miR-217 and commonly overexpressed miR-217 promoted cell cycle progression and enhanced tumor proliferation via inhibiting DACH1 in breast cancer.
According to previous reports, microRNAs were involved in almost every step during the initiation, development and progression of breast cancer[18-20]. Based on their functions, microRNAs could be grouped as onco-miRs (oncogene-like microRNA) and tumor suppressor-miRs (tumor suppressor like microRNA)[21]. Herein, we proved miR-217 as a novel onco-miR in breast cancer, which could accelerate the cell cycle progression via directly targeting the DACH1 gene and inhibit its anti-cancer functions. Up to our knowledge, miR-217 was the first microRNA which directly targeted DACH1.
Figure 4
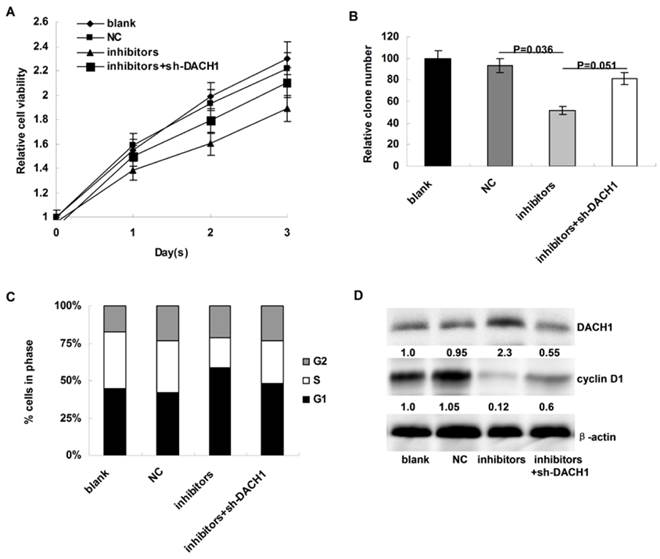
Inhibition of miR-217 suppressed cellular growth and arrested cell cycle via upregulating DACH1 in MDA-MB-231 cells. (A) compared to the controls, miR-217 inhibition notably decelerated cellular growth in MDA-MB-231 cells (P=0.007), which could be partially reversed when DACH1 was pre-inhibited (P=0.033); (B) the colony formation were also suppressed by miR-217 inhibition (P=0.036), which was significantly reversed by the DACH1 pre-inhibition (P=0.051); (C and D) compared to the controls, miR-217 inhibition induced G1 phase arrest and decreased S phase in MDA-MB-468 cells (P=0.027), which was significantly neutralized by the pre-inhibition of DACH1 (P=0.041). The expression of cyclin D1 protein presented similar alterations.
Figure 5
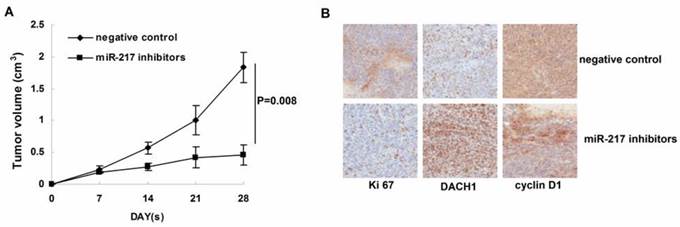
Inhibition of miR-217 suppressed tumor growth in vivo. (A) treatment with miR-217 inhibitors significantly suppressed the growth of MDA-MB-231 xenografts (P=0.008); (B) inhibition of miR-217 upregulated the level of DACH1 while downregulated the expression of Ki 67 and cyclin D1.
Consistent with our findings, miR-217 was demonstrated as an oncogene in aggressive human B-cell lymphomas, which could downregulate the expression of a DNA damage response and repair gene network and in turn stabilizes Bcl-6[22]. On the contrast, several studies supported that miR-217 might serve as a tumor suppressor[23, 24]. In hepatocellular carcinoma, the expression of miR-217 expression was much lower in highly invasive cell lines and metastatic tissues. In vitro and in vivo studies proved that miR-217 could directly target E2F3 and consequentially inhibit tumor invasion[23]. In another study of pancreatic ductal adenocarcinoma, miR-217 was found to be able to inhibit the functions of KRAS and loss of which enhanced tumor proliferation[24]. Considering this discordance was reported in a few kinds of cancers, we hypothesized the roles of miR-217 might be cancer species-dependent, which required further investigations in the future.
Cyclin D1 could manipulate the G1 to S phase during cell cycle through activating the cyclin-dependent kinase 4 and 6 (CDK4/6)[25, 26]. Sequential phosphorylation of the key substrates by the complex of cyclin D1 and CDK4/6 promotes the timely induction of cellular DNA synthesis[27]. Oncogenic disruption of the cell cycle machinery (such as upregulation of cyclin D1 and downregulation of p21/p27) was a common phenomenon in breast cancer[28, 29]. Pestell et al. previously reported that DACH1 could repress the expression of cyclin D1 via a c-Jun DNA-binding partner[8]. In our study, the loss of DACH1 induced by miR-217 significantly unregulated cyclin D1 in breast cancer cells, providing another mechanism of cyclin D1 overexpression in breast cancer.
In summary, we found that miR-217 was commonly overexpressed in breast cancer, which could enhance tumor proliferation via promoting cell cycle progression. Moreover, the DACH1 (the cell fate determination factor) was identified as a novel target of miR-217. Our results proposed inhibiting miR-217 to be a potent therapeutic strategy for breast cancer.
Acknowledgements
Our study was supported by Wu Jieping Medical Foundation (320.6750.14070) and science and Technology Fund of Liaoning Province (20141183).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69-90
2. Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64:9-29
3. Poortmans P, Marsiglia H, De Las Heras M, Algara M. Clinical and technological transition in breast cancer. Rep Pract Oncol Radiother. 2013;18:345-52
4. Rivenbark AG, O'Connor SM, Coleman WB. Molecular and cellular heterogeneity in breast cancer: challenges for personalized medicine. Am J Pathol. 2013;183:1113-24
5. Chen R, Amoui M, Zhang Z, Mardon G. Dachshund and eyes absent proteins form a complex and function synergistically to induce ectopic eye development in Drosophila. Cell. 1997;91:893-903
6. Shen W, Mardon G. Ectopic eye development in Drosophila induced by directed dachshund expression. Development. 1997;124:45-52
7. Sunde JS, Donninger H, Wu K, Johnson ME, Pestell RG, Rose GS. et al. Expression profiling identifies altered expression of genes that contribute to the inhibition of transforming growth factor-beta signaling in ovarian cancer. Cancer Res. 2006;66:8404-12
8. Wu K, Li A, Rao M, Liu M, Dailey V, Yang Y. et al. DACH1 is a cell fate determination factor that inhibits cyclin D1 and breast tumor growth. Mol Cell Biol. 2006;26:7116-29
9. Dalgin GS, Drever M, Williams T, King T, DeLisi C, Liou LS. Identification of novel epigenetic markers for clear cell renal cell carcinoma. J Urol. 2008;180:1126-30
10. Yamada Y, Arao T, Gotoda T, Taniguchi H, Oda I, Shirao K. et al. Identification of prognostic biomarkers in gastric cancer using endoscopic biopsy samples. Cancer Sci. 2008;99:2193-9
11. Wu K, Liu M, Li A, Donninger H, Rao M, Jiao X. et al. Cell fate determination factor DACH1 inhibits c-Jun-induced contact-independent growth. Mol Biol Cell. 2007;18:755-67
12. Wu K, Katiyar S, Li A, Liu M, Ju X, Popov VM. et al. Dachshund inhibits oncogene-induced breast cancer cellular migration and invasion through suppression of interleukin-8. Proc Natl Acad Sci U S A. 2008;105:6924-9
13. Yan W, Wu K, Herman JG, Brock MV, Fuks F, Yang L. et al. Epigenetic regulation of DACH1, a novel Wnt signaling component in colorectal cancer. Epigenetics. 2013;8:1373-83
14. He L, Hannon GJ. MicroRNAs: small RNAs with a big role in gene regulation. Nat Rev Genet. 2004;5:522-31
15. Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281-97
16. Kloosterman WP, Plasterk RH. The diverse functions of microRNAs in animal development and disease. Dev Cell. 2006;11:441-50
17. Nan F, Lu Q, Zhou J, Cheng L, Popov VM, Wei S. et al. Altered expression of DACH1 and cyclin D1 in endometrial cancer. Cancer Biol Ther. 2009;8:1534-9
18. Silveri L, Tilly G, Vilotte JL, Le Provost F. MicroRNA involvement in mammary gland development and breast cancer. Reproduction, nutrition, development. 2006;46:549-56
19. Khoshnaw SM, Green AR, Powe DG, Ellis IO. MicroRNA involvement in the pathogenesis and management of breast cancer. J Clin Pathol. 2009;62:422-8
20. Yu Z, Baserga R, Chen L, Wang C, Lisanti MP, Pestell RG. microRNA, cell cycle, and human breast cancer. Am J Pathol. 2010;176:1058-64
21. Zhang B, Pan X, Cobb GP, Anderson TA. microRNAs as oncogenes and tumor suppressors. Dev Biol. 2007;302:1-12
22. de Yebenes VG, Bartolome-Izquierdo N, Nogales-Cadenas R, Perez-Duran P, Mur SM, Martinez N. et al. miR-217 is an oncogene that enhances the germinal center reaction. Blood. 2014;124:229-39
23. Su J, Wang Q, Liu Y, Zhong M. miR-217 inhibits invasion of hepatocellular carcinoma cells through direct suppression of E2F3. Mol Cell Biochem. 2014;392:289-96
24. Zhao WG, Yu SN, Lu ZH, Ma YH, Gu YM, Chen J. The miR-217 microRNA functions as a potential tumor suppressor in pancreatic ductal adenocarcinoma by targeting KRAS. Carcinogenesis. 2010;31:1726-33
25. Kim JK, Diehl JA. Nuclear cyclin D1: an oncogenic driver in human cancer. J Cell Physiol. 2009;220:292-6
26. Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer. 2009;9:153-66
27. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646-74
28. Velasco-Velazquez MA, Li Z, Casimiro M, Loro E, Homsi N, Pestell RG. Examining the role of cyclin D1 in breast cancer. Future Oncol. 2011;7:753-65
29. Zhang Q, Sakamoto K, Wagner KU. D-type Cyclins are important downstream effectors of cytokine signaling that regulate the proliferation of normal and neoplastic mammary epithelial cells. Mol Cell Endocrinol. 2014;382:583-92
Author contact
![]() Corresponding author: Jiang Daqing, Department of Breast Surgery, Liaoning Cancer Hospital & Institute. Email: jdqlncom. Post Address: No.44 Xiaoheyan Road, Dadong district, Shenyang, Liaoning Province, 110042, China. Telephone: 0086+024-31916207. Fax: 0086+024-31916207
Corresponding author: Jiang Daqing, Department of Breast Surgery, Liaoning Cancer Hospital & Institute. Email: jdqlncom. Post Address: No.44 Xiaoheyan Road, Dadong district, Shenyang, Liaoning Province, 110042, China. Telephone: 0086+024-31916207. Fax: 0086+024-31916207
Citation styles
APA
Zhang, Q., Yuan, Y., Cui, J., Xiao, T., Jiang, D. (2015). MiR-217 Promotes Tumor Proliferation in Breast Cancer via Targeting DACH1. Journal of Cancer, 6(2), 184-191. https://doi.org/10.7150/jca.10822.
ACS
Zhang, Q.; Yuan, Y.; Cui, J.; Xiao, T.; Jiang, D. MiR-217 Promotes Tumor Proliferation in Breast Cancer via Targeting DACH1. J. Cancer 2015, 6 (2), 184-191. DOI: 10.7150/jca.10822.
NLM
Zhang Q, Yuan Y, Cui J, Xiao T, Jiang D. MiR-217 Promotes Tumor Proliferation in Breast Cancer via Targeting DACH1. J Cancer 2015; 6(2):184-191. doi:10.7150/jca.10822. https://www.jcancer.org/v06p0184.htm
CSE
Zhang Q, Yuan Y, Cui J, Xiao T, Jiang D. 2015. MiR-217 Promotes Tumor Proliferation in Breast Cancer via Targeting DACH1. J Cancer. 6(2):184-191.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) License. See http://ivyspring.com/terms for full terms and conditions.