Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2015; 6(2):128-138. doi:10.7150/jca.10787 This issue Cite
Research Paper
The Quantum of Initial Transformed Cells Potentially Modulates the Type of Local Inflammation Mechanism Elicited by Surrounding Normal Epithelial Tissues and Systemic Immune Pattern for Tumor Arrest or Progression
Lawrence Owusu1, 3, Bo Wang2, Yue Du2, Weiling Li 1 ![]() , Yi Xin 1
, Yi Xin 1 ![]()
1. Department of Biotechnology, Dalian Medical University, Dalian 116044, P.R. China.
2. Department of Pathology, Dalian Medical University, Dalian 116044, P.R. China.
3. Department of Biochemistry and Biotechnology, Kwame Nkrumah University of Science and Technology (KNUST), Kumasi, Ghana.
Received 2014-10-11; Accepted 2014-11-20; Published 2015-1-1
Abstract
The immune/ inflammation system potentially serves to arrest, eliminate or promote tumor development. Nonetheless, factors that dictate the choice are not comprehensively known yet. Using a B16/F1 syngeneic wild type model, we evaluated the essentiality of initial transformed cells' density for overt tumor development, the molecular trends of inflammatory mediators in the normal tumor-adjacent epithelial tissues (NTAT), and how such local events may reflect systematically in the host. Overt tumors developed, within an observatory period of at least 45 days and 90 days at most, only in mice inoculated with cancer cells above a limiting threshold of 1× 103 cells. Immunoblots showed early, intense and transient presence of IL-1β, IFN-γ, and both the all-thiol and disulfide forms of HMGB1 in the NTAT of non-tumor bearing mice. However, all-thiol form of HMGB1 and delayed but aberrant IL-6 expression characterized chronic inflammation in tumor bearing hosts. These local epithelial tissue events uniquely reflected in host's systemic cytokines dynamics where stable Th1/Th2 signature (IFN-γ/ IL-4) coupled with early Th1 cells polarization (IL-12/ IL-4) evidenced in non-tumor hosts but highly fluctuating Th1/ Th2 profile in tumor hosts, even before tumors became overt. This hypothesizes that the physical quantum of transformed cells that may either spontaneously arise or accrue at a locus may be crucial in orchestrating the mechanism for the type of local epithelial tissue and systemic immune/ inflammatory responses essential for tumor progression or arrest.
Keywords: Cancer cell density, minimum tumorigenic threshold, inflammation, overt tumor, Th1/Th2/Th17 cytokines.
Introduction
Development of clinically overt tumor is a multi-faceted dynamic process that is initiated from a nascent transformed cell through to the formation of a bulky heterogeneous cancerous cell population at a specific location within the body [1]. All through the process, and beyond cell-intrinsic tumor suppressor mechanisms, the immune system influences several intrinsic and employs extrinsic mechanisms to either completely eliminate, arrest, or educate for immune escape the transformed cells [2, 3]. All things being equal, pre-malignant and malignant cells can be restrained (occult) unless under dysfunctional or weakened host's antitumor mechanisms; as evidenced by increased tumor incidence in immune compromised subjects [4, 5].
One tumorigenesis concept that has generated diverse views is with cancer stem cells (CSC) or tumor-propagating cells (TPC). While some suggest cancer must be initiated by unique and rare cell types [6, 7], others point to the view that cancer, at least some, needs not be driven by such unique cells [8-10]. Clearly, these works are attempting to address an important question about the cell "quality" essential for tumorigenesis. As the investigations continue, a plausible question in an immunocompetent host is "beyond initial quality, could initial quantity be of any significance and how?"
Rudolf Virchow in 1858 proposed that neoplasms and embryogenesis may share common principles and that inflammation and cancer may be linked [11]. During embryogenesis and development, the numbers of a particular precursor cell type within a specified area, called community, are essential for such cells to develop into a unique functional tissue, in what is termed the community effect [12, 13]. One essential factor observed in a Xenopus embryo model, and modeled in silico, to have led to the collapse of community effect was the initial number of cells [14]. Thus, quality and quantity go hand-in-hand in biological systems hence if this could hold and be extended to tumor development, then at some critical cell number of either spontaneously nascent or over-time-accumulated transformed cells, the host's immune defense mechanisms could be effective at eliminating, restraining or promoting such cells, upon being sensed, thus a physico-physiological immune modulation process.
The activation of the innate and subsequently adaptive immune arm is precipitated by “danger signals or alarmins” [15]. One such evolutionary conserved but functionally dynamic molecule is high mobility group box 1 (HMGB1). It has the potential to promote both cell survival and death by regulating multiple signaling pathways, including inflammation and immunity [16]. Its biological and immune functions are dependent on its post-translational modifications (redox or acetylation), and the presence of other biological molecules such as cytokines [17- 19]. This makes HMGB1 an ideal alarmin that may not only initiate and link but also polarize innate and adaptive immune responses [20]. The overall consequence of these host immune/extrinsic defense mechanisms on the initial transformed cells may either promote their death, arrest or progression.
This study employs B16/F1- C57BL/6 transplantation model to evaluate the significance of initial number/ density of transformed cells for overt tumor development, its influence on some innate immune molecular mediators in immediate non-neoplastic epithelial tissues, and how these local tissue events may reflect on the host's systemic immune/inflammation indicators.
Materials and Methods
Mice and Cell line
Six weeks old female C57BL/6 wild-type (WT) mice were obtained from the Specific Pathogen Free (SPF) Animal Center of Dalian Medical University and maintained until 8 weeks old before used. All animal experiments were approved by the Animal Ethics Committee of Animal Care Center, Dalian Medical University, China. B16/F1 melanoma cells (ATCC, number: CRL-6323, USA) were grown in DMEM supplemented with 10% heat-inactivated fetal bovine serum (GIBCO, USA), 0.1% benzyl penicillin and streptomycin (Thermo Scientific, USA). Cells were maintained at 37°C in a humidified atmosphere of 5% CO2.
Transplant model
Subcutaneous implantation of B16/F1 cells was performed in 100 μl of Hank's Balance Salt Solution (HBSS; Thermo Scientific, USA) at the flank of C57BL/6 WT mice with the indicated number of cells (Table 1). Control mice received 100 μl of HBSS without cells. Animals were monitored for tumor growth at 3-day intervals for at least 45 days, and up to 90 days for non-tumor bearing mice. In a tumor bearing animal, tumor depth, maximal width, and maximal perpendicular length were measured with a digital caliper and the tumor volume estimated using the formulae 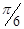 × length × width × depth. The model was repeated three times with similar results.
× length × width × depth. The model was repeated three times with similar results.
Quantum of inoculated B16 cells and tumor incidence in wild type C57BL/6 mice.
| Group | Approx. number of inoculated cells | Tumor incidence a (%) |
|---|---|---|
| C | - | 0/6 (0) |
| G1 | 1 × 102 | 0/15 (0) |
| G2 | 5 × 102 | 0/15 (0) |
| G3 | *1 × 103 | 0/15 (0) |
| G4 | 5 × 103 | 6/15 (40) |
| G5 | 1 × 104 | 12/15 (80) |
| G6 | 5 × 104 | 15/15 (100) |
| G7 | 1 × 105 | 15/ 15 (100) |
C: control which received 100μl of HBSS without cells; * indicates the minimum threshold (MT) of cancer numbers. a Tumor incidence represents the sum of all mice that developed tumors in three independent model experiments of five mice per group in each experiment.
Blood and Tissue sampling
At 45 days post tumor cells implantation, animals were partially sedated by light ether inhalation. Blood samples were collected by one-time orbital sinus bleeding of each animal and subsequently sacrificed by cervical dislocation. Sera were prepared from blood samples, aliquoted and stored at - 80 °C until used. Non-neoplastic epithelial tissues within the immediate surroundings of the tumor bulk, thus normal tumor-adjacent tissue (NTAT), in tumor bearing mice (T) were carefully harvested to avoid contamination from tumor cells or tissue (Fig. 1a). Part of each tissue was immediately frozen or fixed in 4% paraformaldehyde. In control (C) and non-tumor bearing mice (nT), the same procedure was performed to harvest and process epithelial tissues around the sites of injection.
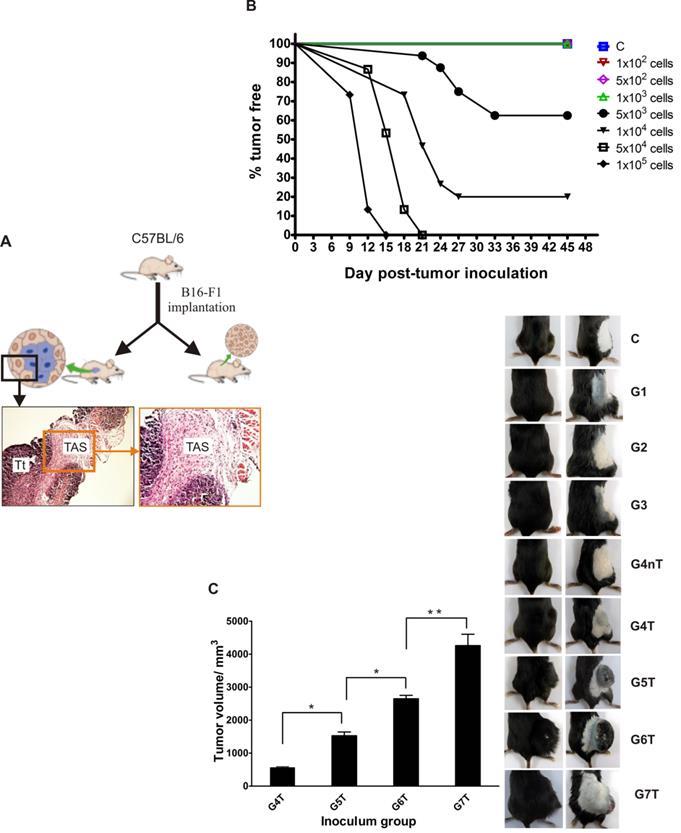
Tumor incidence and average volume are initial tumor inocula dependent: (A) Experimental model schematic. (B) Overt tumor establishment: the higher the dose, the earlier the establishment of tumor. (C) Average tumor volume 6 weeks post-tumor implantation (image and graph). Bar graph data represents mean ± SEM of three independent experiments. Statistical significance was calculated using Student's t test. *p < 0.05, **p < 0.01. C: control; G1: group 1; G2: group 2: G3: group 3; G4: group 4; G5: group 5; G6: group 6; G7: group 7. T: tumor bearing; nT: non-tumor bearing.
Histological analysis
Hematoxylin-eosin (H & E) staining was done on 4 μm sectioned paraffin-embedded specimens that were previously fixed. Briefly, sections were rehydrated and counterstained with hematoxylin for 1 min and eosin for 2 min according to standard protocol. The sections were dehydrated and mounted with cover slips.
Protein extraction, SDS-PAGE and Immunoblotting
Previously harvested tissues were minced and lysed in chilled RIPA lysis buffer (KeyGEN, China) supplemented with protease inhibitor cocktail (Sigma-Aldrich, USA) and PMSF (Thermo Scientific, USA), and protein concentration determined using DC Protein Assay kit (Bio-Rad, USA). Proteins were separated on 12% or 10% bis-tris SDS-polyacrylamide gels under reduced or non-reduced conditions, as detailed elsewhere [21]. Separated proteins were transferred onto 0.2 μm nitrocellulose membrane (Whatman, USA) and probed with primary antibodies overnight at 4ºC, after blocking with 3% BSA in 0.1% PBST. Primary antibodies used were rabbit polyclonal anti -HMGB1, -IL-1β, -IL-6, -IFN-γ (Proteintech, USA), and rabbit anti-VEGF-A (Bioworld, USA). Anti-β-actin (Proteintech, USA) was used to control even sample loading. Membranes were washed thrice in 0.1% PBST for 5 minutes each after primary antibody incubation and the appropriate anti-species HRP-conjugated secondary antibody (Santa Cruz, USA) added for 60 min at room temperature. Protein bands were visualized using Super Signal® West Pico ECL kit (Thermo scientific, USA), and imaged and analyzed with the Gel Documentation and Analysis System (Bio-Rad, USA).
Cytometric bead array (CBA) assay
Serum cytokines including IL-2, IL-4, IL-6, IL-10, TNF-α, IFN-γ and IL-17A (Mouse Th1/Th2/Th17 Cytokine kit), IL-1α, IL-1β, IL-12p70, MCP-1 and GM-CSF were determined using CBA assay (BD Biosciences, USA) according to manufacturer's instructions. Briefly, aliquots of sera were thawed and used undiluted. Beads coated with capture antibodies were mixed or used alone when there was fluorescence position overlap. 50μl of capture bead mixture was added to 50 μl of sample. To this sample-bead mixture, 50 μl of Phycoerythrinconjugated (PE) detection antibody was added and incubated in the dark with occasional shaking. All unbound antibodies were washed prior to beads acquisition on BD FACSCalibur flow cytometer (BD Bioscience, USA). Serial cytokine standards were prepared and treated as above. From the acquired data, standard curves were constructed (R2 > 0.99 for each cytokine) and test calculations performed using FCAP Array Software (BD Biosciences, USA). When the fluorescent signal of a sample was equal to or below that of the positive control, the cytokine level was set as zero.
Statistics
Data were analyzed with GraphPad Prism software (version 5, GraphPad Software Inc., USA) and presented as means ± SEM or median + range, where appropriate, from separate experiments. Significant differences between groups were assessed by Student's t test or Kruskal-Wallis test with Dunn's multiple comparison analysis, where appropriate. P < 0.05 was considered significant.
Results
Overt tumor incidence and volume are cancer cell numbers dependent
We hypothesized that the number of transformed cells (a physico-physiological factor) that could evade immunosurveillance of a host may be essential for tumor development. To explore this, wild-type C57BL/6 mice were inoculated with varying numbers (Table 1) of B16 tumor cells (viability >95% by trypan blue exclusion) and monitored for at least 45 days. All animals inoculated with a thousand tumor cells (minimum threshold), and below, did not develop tumors but above this threshold however, not less than 40% tumor incidence was observed in all mice (Table 1). The time to signs of overt tumor was also tumor-number dependent (Fig. 1B). By day 9 post-implantation, the first tumor was visible in mice that received the highest dose. By day 21, overt tumors were established in some of the mice that had received tumor cells just above the minimum threshold. On day 45 post-implantation, the average tumor volume per group was directly initial-inocula correlated, and significantly differed between groups (Fig. 1C).
Differential microvasculature, IL-6 expression and HMGB1 dynamics in normal tumor-adjacent epithelial tissues (NTAT) of tumor (T) and non-tumor (nT) bearing mice
Endothelial cell proliferation, a sign of angiogenesis, was observed in the NTAT of all tumor bearing, but not non-tumor bearing, animals (Fig. 2A). Western blotting for VEGF-A expression corroborated the histological observation where levels were at least 3.5 fold higher in NTAT of tumor bearing than in non-tumor bearing (nT) mice (Fig. 2B). Interleukin 6 (IL-6) being essential for survival and growth of both normal and premalignant epithelial cells, its expression level was significantly higher in NTAT of tumor bearing mice compared with non-tumor bearing cohorts (Fig. 2C).
Angiogenesis, inflammation and expression pattern of redox forms of HMGB1 in NTAT: (A) To be certain that harvested NTAT were not contaminated with tumor tissues, random sections of harvested NTAT from each group were H & E stained and critically examined under the microscope (Olympus BX41, Japan). Corresponding tumor samples from animals that developed tumors were used for comparison. All NTAT were free from tumor cells/tissue contamination. Endothelial cell proliferation/ angiogenesis (arrows) was common in the NTAT of tumor bearing mice but not in non-tumor or control mice. (B) VEGF-A protein expression levels in NTAT of tumor (T) and non-tumor (nT) bearing mice. Expression levels were quantified by densitometric measurements using ImageJ software. Bars represent mean ± SEM. (C) IL-6 expression in NTAT of tumor (T) bearing and non-tumor (nT) bearing. (D) Visual time course observation of tissue inflammation, tumor arrest or progression in two groups of mice (n = 12 per group) that were either inoculated with 1 × 103 cancer cells (MT) or 5 × 103 cancer cells (Above-MT) and sacrificed at the indicated time points. To aid visualization of inflamed areas and implanted cancer cells, NTAT from MT mice were fixed in 4% paraformaldehyde for 3 days and then visualized on a transluminator (Jim-X Scientific, China). Arrows indicate implanted cells. (E) Time course expression levels of (i) total HMGB1 and (ii) redox forms of HMGB1 in the NTAT of "MT" and "Above-MT" groups. For all Western blot panels, detection of actin served as the loading control. Immunoblots and macrographs are representative of three independent experiments.
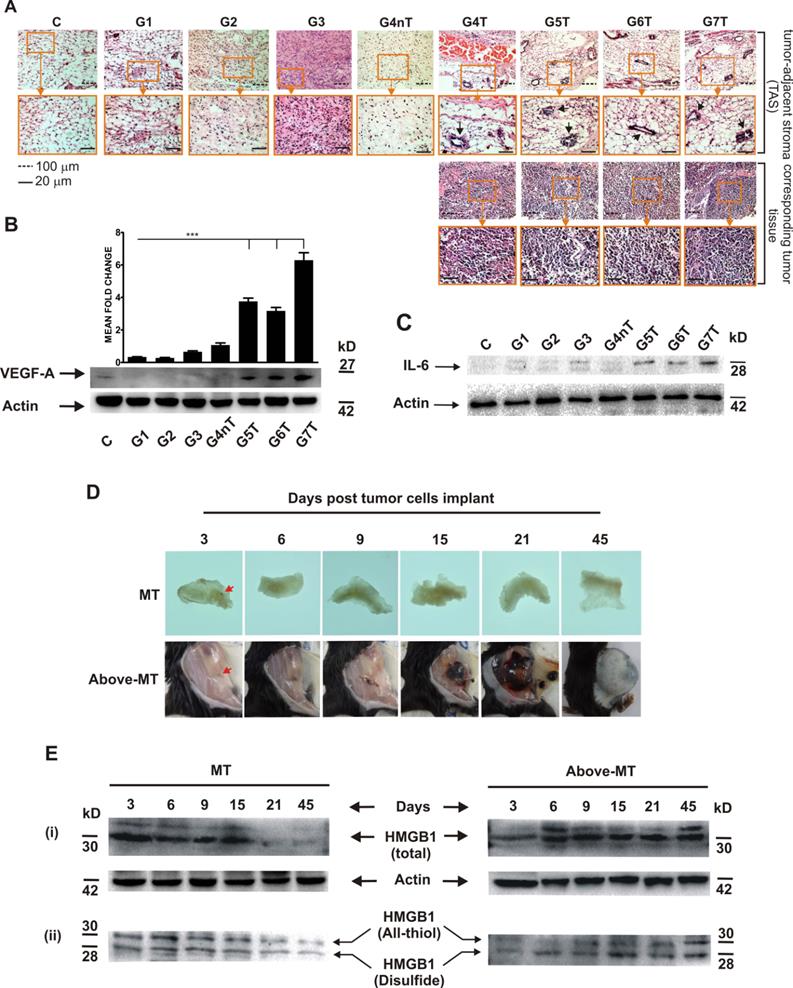
However, to probe the influence of the presence of the different cancer cell densities on inflammation modulation within the immediate normal tissues of the transformed cells, a time series investigation involving two groups of mice that received tumor cells either at the minimum threshold (MT) or higher (Above-MT) was conducted, and their NTAT harvested for analyses. While inoculated cells progressively grew in the "Above-MT" hosts, highly inflamed areas evidently surrounded the inoculated cells in "MT" mice but resolved over time together with the "loss" of the implanted cells (Fig. 2D). Time course expression levels of both total and redox forms of HMGB1 (all-thiol and disulfide) revealed the strong early presence of HMGB1 in the NTAT of non-tumor bearing mice with gradual decreasing levels up to day 15 when the levels were drastically low (Fig. 2E-i). Albeit a relatively lower early induction of HMGB1 in tumor bearing mice, the levels remained constitutively higher from day 6 through to day 45 post-implantation. However, the disulfide form was relatively highly present in tumor bearing mice until latter stages when similar levels of both forms were observed (Fig. 2E-ii).
Expression and dynamics of inflammation mediators differ in the NTAT of tumor bearing (T) and non-tumor bearing (nT) mice
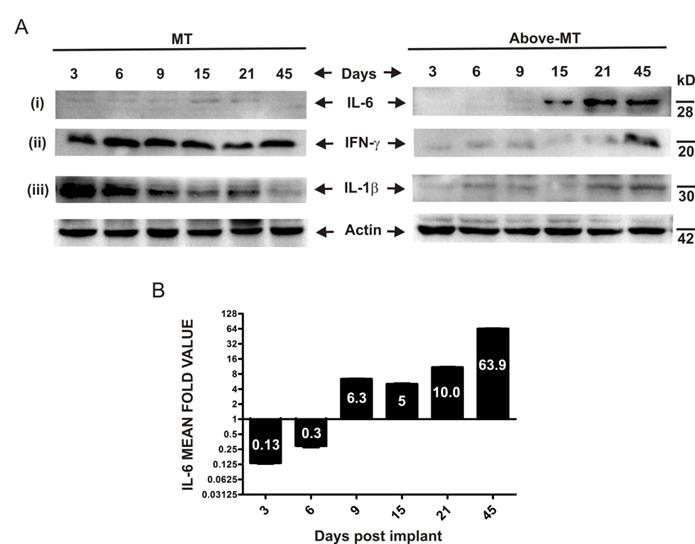
To determine the expression and dynamics of some molecular inflammation mediators, interferon gamma (IFN-γ), interleukin 1 beta (IL-1β) and IL-6 in the NTAT of "MT" and "Above-MT" groups were analyzed by Western blotting. IFN-γ and IL-1β were strongly and significantly present at early stages through to day 21 post-implantation in "MT" mice (Fig. 3A-ii, iii). Conversely, the levels of these mediators were appreciably higher at the very late stage in "Above-MT" cohort. Interestingly, IL-6 was low at the early phase in "MT" mice and remained relatively low throughout the time course (Figs. 3A-i and 3B) but a sharp increase in IL-6, not less than 5 fold, in the NTAT of "Above-MT" mice was observed from the mid through the late stages (Fig. 3A-i and 3B).
Time course expression pattern of inflammation mediators in NTAT of mice: (A) Representative immunoblots of tissue lysates from "MT" and "Above-MT" mice at the indicated time points for (i) IL-6, (ii) IFN-ɣ and (iii) IL-1β expression levels. Actin was used as loading control. (B) Bar graph of densitometric analysis of IL-6 fold change of corresponding ratios of "Above-MT" to "MT" during the time course. Data represent mean ± SEM from three independent analyses.
Systemic profile and dynamics of Th1/Th2/Th17, Proinflammatory, MCP-1 and GM-CSF cytokines in MT and Above-MT mice
To evaluate how the initial transformed cells' progression or elimination could systemically reflect on the immune/inflammatory machinery, an end-point (Suppl. 2) and time course Th1/Th2/Th17, IL-α and IL-β, monocyte chemotactic protein-1 (MCP-1) and granulocyte-monocyte colony stimulation factor (GM-CSF) cytokines were assayed (Fig. 4). Serum samples obtained from "MT" and "Above-MT" mice at the indicated time points were used for the time series study. In mice without tumors, serum Th1 cytokines (IL-2, IFN-γ, TNF-α and IL-12) levels were generally higher, at least, during the early (3- 6 days post-implantation) phase of the process (Fig. 4A). Surprisingly, Th2 cytokine levels were also generally high in "MT" compared with tumor bearing mice (Fig. 4A). IL-10, a regulatory cytokine (like IL-17A, Fig. 4C), was higher at the earliest point in the process in "Above-MT" group but declined and remained low compared with levels in "MT" cohort.
Systemic cytokinomics of "MT" and "Above-MT" mice: (A) Th1 cytokines. (B) Th2 cytokines. (C) IL-17. (D) Th1/ Th2 stability. (E) IL-1α and IL-1β. (F) MCP-1 and GM-CSF. Data represent median ± range.
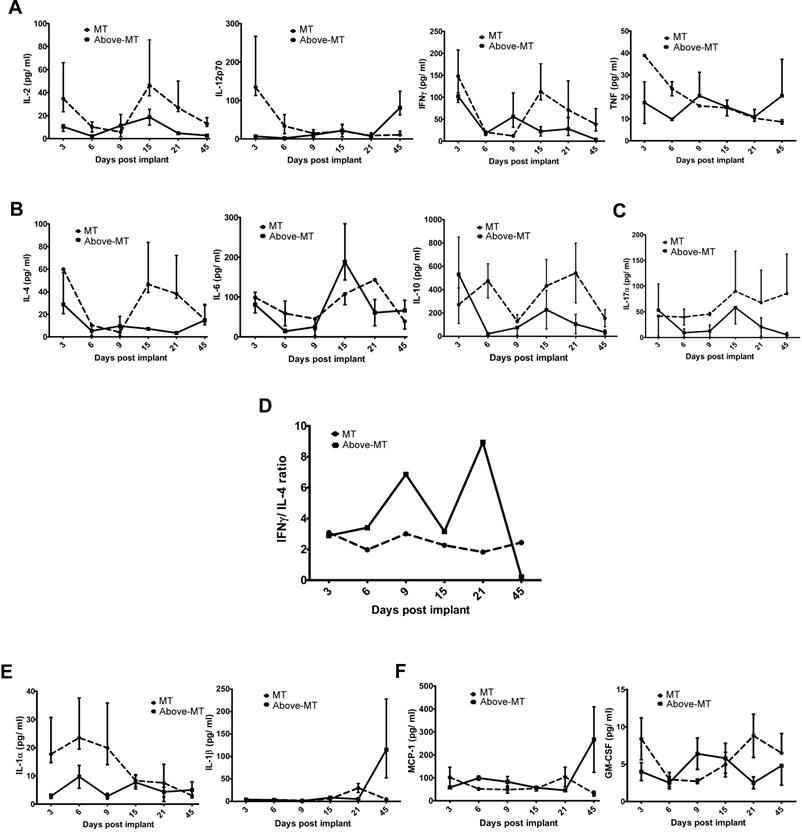
Th1/ Th2 stability using systemic IFN-γ/ IL-4 scores were determined. Intriguingly, a highly fluctuating Th1/ Th2 signature was observed in the "Above-MT" group, even before tumors became overt, while a fairly stable status was observed in the "MT" group (Fig. 4D). An indicated inverse trend of Th1/Th2 cell polarization (IL-12/ IL-4 ratios) of dominant Th1 was associated with "MT" group from the early to the mid phase of the process (Suppl. 3).
Evaluation of the proinflammatory cytokines IL-1α and IL-1β revealed the former to be elevated in circulation at early to mid-phase in "MT" than in "Above-MT" group but its level waned while IL-1β, though initially low, sharply increased in the "Above-MT" group at the late phase, thus after tumor development (Fig. 4E). MCP-1 levels were steadily low at the early and mid-phases in both groups but sharply increased at the late phase in "Above-MT" group, a possible indication of increased monocytes trafficking and infiltration (Fig. 4F). In "MT" mice, GM-CSF was higher in circulation at early and late phases but a direct inverse trend was observed in "Above-MT" mice (Fig. 4F).
Discussion
In an immunocompetent host, the immune system is not silent but serves as a potent extrinsic antitumor machinery. It is capable of completely eliminating and/ or keeping in dormant state (equilibrium) nascent transformed cells [3, 22]. Currently, it is accepted that through some of its effector processes, such as inflammation, the immune system aids in the tumor escape-progression process [23]. However, it remains largely unclear what the initial critical deterministic factors and events could be to switch immunity either for or against tumor progression. Aside most cancers arising from the epithelia, current studies point also to key roles of epithelial cells in inflammation which include taking up of apoptotic cells, and cytokines secretion to regulate immune responses [40, 41]. We used parental B16/F1 cells, which are generally poorly immunogenic but aggressively form tumors in syngeneic mice [24], as rough representation of post-immunoedited transformed cells to investigate the likely significance of cancer cell "quantity" in overt tumor development in an immunocompetent host.
The data prompts that there could be a possible minimum load of cancer cells that must be present within a limited area to promote the establishment of an overt tumor (Table 1). This observation fits the community effect phenomenon where interactions among nearby precursor cells enable them to maintain tissue-specific gene expression, secrete critical molecules and differentiate in a coordinated manner [12, 13]. Within the limitations of the current model, it was evidenced that in immunocompetent mice overt tumors could develop only when the initial transplanted viable cells were above a minimum number of 1× 103 cells and that at and below this threshold, mice did not develop tumors within the reported period (Table 1 and Fig. 1B), and even up to 90 days (data not shown). Also, the higher the initial tumor cells, the earlier the tumor onset and the larger the average tumor volume (Fig. 1B and 1C). In a recent in silico study, Saka et al. [14] using a combination of stochastic and deterministic modeling theoretically arrived at a similar conclusion that a critical initial number of cells were essential for the initiation and maintenance of a steady-state community effect. These observations may suggest that the number or density of transformed cells that may spontaneously escape immunosurveillance or that may accumulate over a period of time at a focus until above a limiting threshold may be essential in the establishment of overt tumor.
Thriving of the initiated tumor would require constant and adequate supply of nutrients and oxygen, and therefore the need to augment microvessel density [25]. The abundant blood vessels and significant expression of VEGF-A, the key angiogenesis driver, might have served this purpose in tumor bearing mice (Fig. 2A and 2B). VEGF-A, in addition to inducing angiogenesis, can promote vascular permeability, vasculogenesis, and inhibit apoptosis of transformed cell [25]. As a proinflammatory and protumor cytokine, the abundant local presence of IL-6 (Fig. 2C) could promote and link chronic inflammation with endothelial proliferation to induce angiogenic switch [26]. Therefore, a "Strength in unity" phenomenon could be operating whereby transformed cells once above a critical threshold were able to "over power" or subvert host's local extrinsic antitumor control measures to progress onto overt.
Local inflammation is one of the early innate immune responses generated during cell/ tissue stress. The ensuing outcome could be beneficial or detrimental based on several factors within the inflammatory microenvironment [11, 27, 28]. In the light of this paradoxical role played by inflammation, we investigated the interactions between the tumors (or tumor cells) and their immediate normal tissue in a time course. Areas surrounding the sites of tumor implantation in "MT" mice were highly inflamed around the inoculated tumor cells (Fig. 2D, arrowed). The inflammation eventually resolved coupled with the "loss" of the implanted transformed cells which, speculatively, might have been cleared upon their death (they were not specifically traced in this study). The early and higher presence of the danger signal, HMGB1, and equally abundant presence of its immune stimulatory disulfide form [29] at the transformed cells' local site in non-tumor bearing mice (Fig. 2E) may have initiated the quick recruitment to and stimulation of innate immune cells at the local area of the transformed cells, thus an acute inflammatory response. The source of HMGB1 could be from the death of transformed cells, due to lack of steady-state community effect, and/ or active secretion from recruited innate immune cells [30, 31].
Immune/ inflammatory cells, particularly dendritic cells (DCs) and monocytes/macrophages, are initially polarized prior to their migration to secondary lymphoid organs based on the cytokine conditions at the site of innate immune response. In the presence of Th1 cytokines, these antigen presenting cells (APCs) mature and polarize into type-1 phenotypes which are very efficient at inducing cell-mediated immunity, and simultaneously secreting high levels of other type 1 cytokines [32- 34]. This could be the situation in the NTAT of "MT" mice where there was an abundant and early presence of IFN-γ, the chief Th1 cytokine (Fig. 3A-ii). Also, HMGB1 is known to bind to appropriate receptors and activate these APCs. For instance, released HMGB1 binds to Toll-like receptor 4 (TLR-4) on DCs and activate and stimulate the processing of tumor antigens for optimal presentation to the adaptive arm of host immunity [35]. Significant to this process within the APC is inflammasome activation which in turn process and facilitate the secretion of IL-1β, a critical signaling molecule mandatory for priming IFN-γ-producing cells and cytotoxic T lymphocytes (CTLs) [36, 37]. Therefore, the early and strong presence of both all-thiol and disulfide forms of HMGB1 (chemotactic and stimulatory, respectively) coupled with the early higher level of IL-1β in the NTAT of "MT" mice (Figs. 2E-ii and 3A-ii, iii) may have promoted timely recruitment of immune cells, their effective activation, IFN-γ secretion and coordinated innate and adaptive antitumor immune responses against the "struggling-to-survive" and possibly dying transformed cells for their consequential elimination. On the contrary, sustained expression and secretion of IL-6 within tumors and surrounding tissues is a hallmark of tumor-promoting inflammation by enhancing the survival and growth of both normal and premalignant epithelial cells [38]. The very high IL-6 expression (together with STAT3 activation and constitutive NFκB expression, Suppl. 1) at the mid-through-late phase (Figs. 3A-i and 3B), coupled with the very low initial IL-1β and IFN-γ but constitutive presence of cytokine-like disulfide HMGB1(Fig. 2E-ii) in "Above-MT" hosts may have led to the lack of or impaired early inflammasome activation, and possibly type-2 phenotype polarization of APCs which predominantly exhibit poor antigen-presenting ability, produce factors that suppress T cell expansion, but promote angiogenesis and sustain chronic inflammation [26]. Notable also, was the relatively higher late phase expressions of IL-1β and IFN-γ in NTAT of tumor bearing mice (Fig. 3A-ii, iii). These were ineffective to contain the tumor bulk, once established. Many tumors, including breast cancer and advanced melanomas, persistently produce IL-1β [39] and therefore the late presence of IL-1β and IFN-γ may be tumor-engineered and as proinflammatory molecules, may contribute to the maintenance of the chronic inflammatory milieu.
The local immune/ inflammation processes against the different load of transformed cells uniquely reflected on the general immunity indicators of the hosts. Generally, early induction and higher levels of circulating Th1 cytokines were evidenced in non-tumor and control mice than their tumor bearing counterparts (Suppl. 2, Fig. 4A). More importantly, the effectiveness of this extrinsic antitumor mechanism appeared to be time and stability dependent (Fig. 4D). This stability could be an index of antitumor immune robustness. However, intense systemic fluctuations [42] in this stability may be indicative of the immune system under stress due to ongoing local contention between tumor cells/ community and the immune machinery, thus providing a permissive context for tumor escape and progression. In the treatment of superficial bladder cancer by Bacillus Calmette-Guerin (BCG) administration, the underlying mechanism for its clinical efficiency is a robust Th1 induction and immune response [43]. De Boer et al. [44] reported that the boosted Th1 response was essentially BCG-viability (quality) as well as BCG-concentration (quantity) dependent. Our model demonstrates that viable transformed cells (quality) are also essential for the induction of Th1 response but on the contrary, the stability and efficiency of the response require the density of the transformed cells to be below a minimum threshold (Fig. 4D). This proposes an inverse mode of effective Th1 response induction using microorganisms [45] or cancer cells and to buttress this, small numbers of viable residual tumor cells were reported to be critical for the initiation and sustenance of concomitant immunity in wild-type hosts [46], thus emphasizing the significant role that physico-physiological factors such as the initial number (quantity) of "foreign body" may play in scalping immunity, and in these cases host's antitumor immunity.
IL-17 is proinflammatory and together with other cytokines (IL-6, IL-1 and TNF-α) and chemokines orchestrates tissue inflammation to recruit Th1 cells to target tissues. Its activity also attracts functional regulatory T cells (Tregs), hence may mediate effector T cells regulation, in the absence of other overwhelming proinflammatory cytokines such as IL-6 [32]. Such plastic function of IL-17 (Fig. 4C) may have occurred whereby it functioned as proinflammatory, in conjunction with elevated levels of IL-1α, TNF-α and IL-6 at the early phase, but regulatory in the presence of lower levels of these cytokines, but higher level of IL-10 (Figs. 4A, 4B and 4E) in "MT" mice to resolve the initial intense inflammation (acute) which could be tissue damaging if sustained.
MCP-1 and GM-CSF serum levels was indicative of increased monocytes trafficking and (rising) stimulation at the late phase in "Above-MT" group as compared to their non-tumor bearing cohort (Fig. 4F). This event is likely protumor/ tumor-driven to infiltrate the tumor and its microenvironment with immune/ inflammatory cells essential for the maintenance of a chronic and tumor-promoting milieu [27].
In summary, the data suggest that in addition to the quality of intrinsic aberrations of cancer cells, there may be an essential minimum transformed cell mass that influence elicited mechanism of local epithelial tissue inflammation in an immunocompetent host during tumorigenesis. The tendency for acute or chronic inflammation in the immediate normal tissue of transformed cells was, at least in part, modulated by the initial number of transformed cells at a locus which eventually influenced the systemic innate immune/ inflammation cytokine patterns and signatures, even before tumors became overt in the host. However, whether our findings are restricted to the current model or have more universal applicability call for investigations using other syngeneic models.
Abbreviations
NTAT: normal tumor-adjacent tissue; HMGB1: high mobility group box protein 1; IL: interleukin; VEGF-A: vascular endothelial growth factor-A.
Acknowledgements
We thank Dr. Benjamin Arko-Boham and Dr. Max Annani Akollor for proofreading the manuscript prior to submission.
Conflict of Interest
The authors declare no conflict of interest.
References
1. Kumar V, Abbas AK, Fausto N. Neoplasia. In: (ed.) Kumar V, Abbas AK, Fausto N. Robbins and Cotran pathologic basis of disease, 7th ed. New Delhi: Elsevier Inc. 2004:276-277 309
2. Zitvogel L, Tesniere A, Kroemer G. Cancer despite immunosurveillance: immunoselection and immunosubversion. Nat Rev Immunol. 2006;6:715-727 doi:10.1038/nri1936
3. Vesely MD, Kershaw MH, Schreiber RD, Smyth MJ. Natural innate and adaptive immunity to cancer. Annu Rev Immunol. 2011;29:235-271 doi: 10.1146/annurev-immunol-031210-101324
4. Deeken JF, Tjen-A-Looi A, Rudek MA, Okuliar C, Young M, Little RF, Dezube BJ. The rising challenge of non-AIDS-defining cancers in HIV-infected patients. Clin Infect Dis. 2012;55:1228-1235 doi: 10.1093/cid/cis613
5. Campisi J. Aging, Cellular Senescence, and Cancer. Annu Rev Physiol. 2013;75:685-705 doi: 10.1146/annurev-physiol-030212-183653
6. Schatton T, Murphy GF, Frank NY. et al. Identification of cells initiating human melanomas. Nature. 2008;451:345-349 doi: 10.1038/nature06489
7. Ishizawa K, Rasheed ZA, Karisch R. et al. Tumor-Initiating Cells are rare in many human tumors. Cell Stem Cell. 2010;7:279-282 doi: 10.1016/j.stem.2010.08.009
8. Adams JM, Kelly PN, Dakic A, Stephen L. Nutt SL, Andreas Strasser. Response to Comment on “Tumor growth need not be driven by rare cancer stem cells”. Science. 2007;318:1722-1723 doi: 10.1126/science.1149672
9. Held MA, Curley DP, Dankort D, McMahon M, Muthusamy V, Bosenberg MW. Characterization of melanoma cells capable of propagating tumors from a single cell. Cancer Res. 2010;70:388-397 doi: 10.1158/0008-5472.CAN-09-2153
10. Huang SD, Yuan Y, Tang H, Liu XH, Fu CG, Cheng HZ, Bi JW, Yu YW, Gong DJ, Zhang W, Chen J, Xu ZY. Tumor cells positive and negative for the common cancer stem cell markers are capable of initiating tumor growth and generating both progenies. PLoS One. 2013;8:e54579. doi: 10.1371/journal.pone.0054579
11. Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow? Lancet. 2001;357:539-545 doi: 10.1016/S0140-6736(00)04046-0
12. Gurdon JB. A community effect in animal development. Nature. 1988;336:772-774 doi:10.1038/336772a0
13. Gurdon JB, Tiller E, Roberts J, Kato K. A community effect in muscle development. Curr Biol. 1993;3:1-11 doi: 10.1016/0960-9822(93)90139-F
14. Saka Y, Lhoussaine C, Kuttler C, Ullner E, Thiel M. Theoretical basis of the community effect in development. BMC Syst Biol. 2011;5:54-68 doi: 10.1186/1752-0509-5-54
15. Gallucci S, Matzinger P. Danger signals: SOS to the immune system. Curr Opin Immunol. 2001;13:114-119 doi: 10.1016/S0952-7915(00)00191-6
16. Kang R, Zhang Q, Zeh HJ 3rd, Lotze MT, Tang D. HMGB1 in Cancer: Good, Bad, or Both? Clin Cancer Res. 2013;19:4046-4057 doi: 10.1158/1078-0432.CCR-13-0495
17. Tang D, Kang R, Cheh CW, Livesey KM, Liang X, Schapiro NE, Benschop R, Sparvero LJ, Amoscato AA, Tracey KJ, Zeh HJ, Lotze MT. HMGB1 Release and Redox Regulates Autophagy and Apoptosis in Cancer Cells. Oncogene. 2010;29:5299-5310 doi: 10.1038/onc.2010.261
18. Yang H, Antoine DJ, Andersson U, Tracey KJ. The many faces of HMGB1: molecular structure-functional activity in inflammation, apoptosis, and chemotaxis. J Leukoc Biol. 2013;93:865-873 doi: 10.1189/jlb.1212662
19. Bianchi ME. HMGB1 loves company. J Leukoc Biol. 2009;86:573-576 doi: 10.1189/jlb.1008585
20. Bianchi ME, Manfredi AA. High-mobility group box 1 (HMGB1) protein at the crossroads between innate and adaptive immunity. Immunol Rev. 2007;220:35-46 doi: 10.1111/j.1600-065X.2007.00574.x
21. Garfin DE. Gel electrophoresis of proteins. Davey J, Lord M, eds. Essential Cell Biology, Oxford: Oxford Univ Press. 2003:2-19
22. Koebel CM, Vermi W, Swann JB, Zerafa N, Rodig SJ, Old LJ, Smyth MJ, Schreiber RD. Adaptive immunity maintains occult cancer in an equilibrium state. Nature. 2007;450:903-907 doi:10.1038/nature06309
23. de Visser KE, Eichten A, Coussens LM. Paradoxical roles of the immune system during cancer development. Nat Rev Cancer. 2006;6:24-37 doi:10.1038/nrc1782
24. Murakami T, Cardones AR, Finkelstein SE, Restifo NP, Klaunberg BA, Nestle FO, Castillo SS, Dennis PA, Hwang ST. Immune evasion by murine melanoma mediated through CC Chemokine Receptor-10. J Exp Med. 2003;198:1337-1347 doi: 10.1084/jem.20030593
25. Hanahan D, Weinburg RA. Hallmarks of Cancer: The Next Generation. Cell. 2011;144:646-674 doi: 10.1016/j.cell.2011.02.013
26. Lewis CE, Pollard JW. Distinct role of macrophages in different tumor microenvironments. Cancer Res. 2006;66:605-612 doi: 10.1158/0008-5472.CAN-05-4005
27. Grivennikov SI, Greten FR, Karin M. Immunity, Inflammation and Cancer. Cell. 2010;140:883-899 doi: 10.1016/j.cell.2010.01.025
28. Rodriguez-Vita J, Lawrence T. The resolution of inflammation and cancer. Cytokine Growth Factor Rev. 2010;21:61-65 doi: 10.1016/j.cytogfr.2009.11.006
29. Venereau E, Casalgrandi M, Schiraldi M. et al. Mutually exclusive redox forms of HMGB1 promote cell recruitment or pro-inflammatory cytokine release. J Exp Med. 2012;209:1519-1528 doi: 10.1084/jem.20120189
30. Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. 2002;418:191-195 doi:10.1038/nature00858
31. Gardella S, Andrei C, Ferrera D, Lotti LV, Torrisi MR, Bianchi ME, Rubartelli A. The nuclear protein HMGB1 is secreted by monocytes via a non-classical, vesicle-mediated secretory pathway. EMBO Rep. 2002;3:995-1001 doi: 10.1093/embo-reports/kvf198
32. Dardalhon V, Korn T, Kuchroo VK, Anderson AC. Role of Th1 and Th17 cells in organ-specific autoimmunity. J Autoimmun. 2008;31:252-256 doi: 10.1016/j.jaut.2008.04.017
33. Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002;23:549-555 doi: 10.1016/S1471-4906(02)02302-5
34. Becker Y. Molecular Immunological Approaches to Biotherapy of Human Cancers - A Review, Hypothesis and Implications. Anticancer Res. 2006;26:1113-1134
35. Apetoh L, Ghiringhelli F, Tesniere A. et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med. 2007;13:1050-1059 doi: 10.1038/nm1622
36. Franchi L, Eigenbrod T, Muñoz-Planillo R, Nuñez G. The inflammasome: a caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat Immunol. 2009;10:241-247 doi: 10.1038/ni.1703
37. Ghiringhelli F, Apetoh L, Tesniere A. et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat Med. 2009;15:1170-1178 doi: 10.1038/nm.2028
38. Grivennikov SI, Karin M. Autocrine IL-6 signaling: a key event in tumorigenesis? Cancer Cell. 2008;13:7-9 doi: 10.1016/j.ccr.2007.12.020
39. Okamoto M, Liu W, Luo Y, Tanaka A, Cai X, Norris DA, Dinarello CA, Fujita M. Constitutively active inflammasome in human melanoma cells mediating autoinflammation via caspase-1 processing and secretion of interleukin-1β. J Biol Chem. 2010;285:6477-6488 doi: 10.1074/jbc.M109.064907
40. Olszak T, Neves JF, Dowds CM. et al. Protective mucosal immunity mediated by epithelial CD1d and IL-10. Nature. 2014;509:497-502 doi:10.1038/nature13150
41. Juncadella IJ, Kadl A, Sharma AK, Shim YM, Hochreiter-Hufford A, Borish L, Ravichandran KS. Apoptotic cell clearance by bronchial epithelial cells critically influences airway inflammation. Nature. 2013;493:547-551 doi:10.1038/nature11714
42. Burkholder B, Huang RY, Burgess R, Luo S, Jones VS, Zhang W, Lv ZQ, Gao CY, Wang BL, Zhang YM, Huang RP. Tumor-induced perturbations of cytokines and immune cell networks. Biochim Biophys Acta. 2014;1845:182-201 doi: 10.1016/j.bbcan.2014.01.004
43. Ratliff TL. Mechanisms of action of BCG in superficial bladder cancer. Prog Clin Biol Res. 1992;378:103-109
44. De Boer EC, Rooijakkers SJ, Schamhart DH, Kurth KH. Cytokine gene expression in a mouse model: the first instillations with viable Bacillus Calmette-Guerin determine the succeeding Th1 response. J Urol. 2003;170:2004-8 doi: 10.1097/01.ju.0000091826.83705.79
45. Ahmad F, Mani J, Kumar P, Haridas S, Upadhyay P, Bhaskar S. Activation of Anti-Tumor Immune Response and Reduction of Regulatory T Cells with Mycobacterium indicus pranii (MIP) Therapy in Tumor Bearing Mice. PLoS ONE. 2011;6:e25424. doi:10.1371/journal.pone.0025424
46. Kakinuma T, Nadiminti H, Lonsdorf AS, Murakami T, Perez BA, Kobayashi H, Finkelstein SE, Pothiawala G, Belkaid Y, Hwang ST. Small numbers of residual tumor cells at the site of primary inoculation are critical for anti-tumor immunity following challenge at a secondary location. Cancer Immunol Immunother. 2007;56:1119-1131 doi: 10.1007/s00262-006-0253-4
Author contact
![]() Corresponding authors: Xin Yi and Weiling Li, Department of Biotechnology, Dalian Medical University, 9 West Section, Lvshun South Road, Dalian, Liaoning 116044, P.R. China. E-mail: jimxincom and liweiling2006com.
Corresponding authors: Xin Yi and Weiling Li, Department of Biotechnology, Dalian Medical University, 9 West Section, Lvshun South Road, Dalian, Liaoning 116044, P.R. China. E-mail: jimxincom and liweiling2006com.