Journal of Cancer
ISSN: 1837-9664
3.2
Impact Factor
ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2015; 6(1):48-53. doi:10.7150/jca.10521 This issue Cite
Research Paper
Upregulation of Acetylcholinesterase Mediated by p53 Contributes to Cisplatin-Induced Apoptosis in Human Breast Cancer Cell
Xiaolei Ye1*, Changsong Zhang2*, Yichen Chen1, Tianbao Zhou3 ![]()
1. Ningbo Institute of Medical Sciences, Ningbo 315020, China
2. Clinical Oncology Laboratory, Changzhou Cancer Hospital of Soochow University, Changzhou 213002, China
3. The Hepatobiliary Surgery Centre, the Ningbo No. 2 Hospital, Ningbo 315010, China
*These authors contributed equally to this work.
Received 2014-9-10; Accepted 2014-10-21; Published 2015-1-1
Citation:
Ye X, Zhang C, Chen Y, Zhou T. Upregulation of Acetylcholinesterase Mediated by p53 Contributes to Cisplatin-Induced Apoptosis in Human Breast Cancer Cell. J Cancer 2015; 6(1):48-53. doi:10.7150/jca.10521. https://www.jcancer.org/v06p0048.htm
Other stylesAbstract
Background: The expression of acetylcholinesterase (AChE) could be induced during apoptosis in various cell types. And reduced AChE expression either by siRNA could prevent apoptosis. However, the detailed mechanisms underlying the AChE regulation are largely unknown in human breast cancer cell.
Material and methods: MCF-7 cells were cultured and treated by cisplatin in the absence or presence of p53 siRNA.
Results: In this study, the regulation of AChE expression during apoptosis induced by cisplatin, a current used anticancer drug, was investigated in human breast cancer cell line MCF-7. Exposure of MCF-7 cells to cisplatin resulted in apoptosis in a time- and concentration-dependent manner. Meanwhile, the upregulated AChE and p53 were also observed during apoptosis. Silencing interfering RNA directed against p53 blocked the expression of AChE.
Conclusion: Taken together, these results suggested that AChE expression could be upregulated by the activation of p53 during apoptosis induced by cisplatin in MCF-7 cells.
Keywords: acetylcholinesterase, apoptosis, p53, cisplatin, breast cancer.
Introduction
Acetylcholinesterase (AChE) plays a significant role in hydrolysis of acetylcholine to block neurotransmission at cholinergic synapses [1]. The AChE is encoded by a single gene. And there are three different isoforms: AChE-S, AChE-E and AChE-R, according to existence of alternative splicing at C-terminus of AChE mRNA [2, 3]. Generally, AChE expression is only found in tissues which are innervated by cholinergic nerves [4]. However, recent studies have reported that AChE was also expressed in several types of hematopoietic cells including erythrocytes and megakaryocytes [5]. It had reported that the elicitation of AChE-S expression was found in different cell types involving non-muscle, non-nervous and non-hematopoietic cell lines [6]. AChE also plays a role in tumorigenesis, which are amplified, mutated, and/or aberrantly expressed in a variety of human tumor types. Cholinesterase activity and acetylcholinesterase glycosylation are altered in human breast cancer [7, 8]. These changes could be the result of chromosome breakage in such as hemopoietic malignancies, which shows that AChE may be involved in apoptosis [9].
P53, as a tumor suppressor, its major function has been changed to pay more attention on cell cycle arrest and apoptosis [10, 11]. It is very important for us to understanding the mechanism by which p53 induces apoptosis, which could be a new anticancer therapeutics in future [12]. p53 could increase mitochondrial outer membrane permeabilization by Bcl-2 family proteins, which implied the mechanism of apoptosis by stress-induced [13].
The high expression of AChE could inhibit proliferation and promote apoptosis in NRK cells [14]. The siRNA AChE could silence myoblasts and high expression of AChE could increase myoblasts, which implied AChE play an important role in apoptosis [15]. The report implied that AChE is very important regulator for cell proliferation and the drug sensitivity in HCC cells [16]. In addition, the detailed mechanisms underlying the AChE regulation in apoptosis are largely unclear.
Increasing studies have demonstrated that AChE and p53 are associated with cell apoptosis, respectively, but few reports described that the relationship between AChE and p53 during the cell apoptotic pathway. In this study, we focused on the AChE expression during apoptosis induced by cisplatin to investigate the relation of AChE and p53 in human breast cancer cell line MCF-7.
Materials and Methods
Cell culture
MCF-7 cells (Shanghai Institutes for Biological Sciences, Shanghai, China) were cultured in incubator with 5% CO2 using plastic Petri dished (100mm) in Dulbecco's modified Eagle's medium (Invitrogen China Ltd., Beijing, China) with phenol red, L-glutamine(2mM), penicillin(100U/ml), streptomycin(100μg/ml), gentamicin(50μg/ml), insulin(6ng/ml), hydrocortisone(3.75 ng/ml) and 5% fetal calf serum. Cells medium changed every 2-3 days.
Cisplatin treatment and cell viability detection
The chemotherapeutic agents: cisplatin was purchased from Qilu Pharmaceutical Co., Ltd. (JiNan, Shandong, China). Apoptosis of MCF-7 cells were induced by cisplatin with different concentration: 50μM, 100μM, 150μM, 200μM and 250μM, and collected the cells in 24hours. Meanwhile, another group of MCF-7 was treated with 100μM cisplatin for different time, and the cell viability was calculated in 6h, 12h, 18h, and 24h. Cell viability was determined by MTT assay. Breast cancer cells were seeded in 96-well plates and cultured in the medium. MTT was dissolved PBS at 5 mg/ml and then added into each well. The cell viability in each well was examined by a MTT colorimetric assay. The optical density (OD) value of each sample was measured using a plate reader. The data was expressed as mean ± SD. Collected cells were stained with Hoechst 33342 (10 μM) for 3 min and analysed under a fluorescence microscope with excitation at UV (360 nm).
Western blot analysis
After treatment with 100μM cisplatin, equal numbers of MCF-7 cells were lysed in lysis buffer composed of 0.6M Tris-HCl (PH 6.8), 10% SDS and protease inhibitor cocktail. Samples were incubated at 4℃ 10mins and then centrifuged at 10,000×g for 15mins at 4℃. The supernatants were transferred, mixed and boiled in sample buffer. The supernatants were separated by polyacrylamide gel electrophoresis and transferred to a PVDF membrane (Bio-rad, Hercules, CA, USA). And then incubated the membrane at room temperature in blocking buffer consist of 5% fat-free milk dissolved in 1×TBST(10mM Tris-Base, PH7.5, 100mM NaCl, and 1% Tween 20) for 1h followed by incubation with the blocking buffer containing first antibody, such as Anti-P53 antibody, Anti-Histone H2A antibody (Abcam, Shanghai, China), Anti-AChE antibody, Anti-mouse antibody, Anti-β-actin antibody (Santa Cruze Biotechnology Co., Ltd. Shanghai, China) at 4℃ overnight. The membrane was washed by TBS, then incubating with second antibody for 1h at room temperate. The blot was exposed to ECL blotting system after TBS washing.
AChE immunofluorescence
MCF-7 cells were growth on steriled coverslips in 24-well plates, confirmed the confluence of cells up to 60% before experiment. Use PBS (500μl) to wash the cells twice for each well. And then added 4% paraformaldehyde (250μl) to each well, fixed them 15min at room temperature. Removed paraformaldehyde, and washed cells with PBS (500μl) containing 3%FBS three times. Each well added DPBS(250μl) including 0.2% Triton X-100 for 5min, washed cells by PBS containing 3%FBS three times. Blocked cells with DPBS (500μl) containing 3%FBS and 0.5% Tween-20 for 1h. Replaced blocking buffer with fresh one added first antibody, incubated for 1h at room temperate. Washed cells and added second fluoresce-antibody (Invitrogen China Ltd., Beijing, China) for 30min. Washed cells three times again with PBS containing 3%FBS. Added a drop of anti-fadeometer on each coverslip, took out the coverslips from 24-well plates and put on the glass slide, left it without light for 5min, cells were observed under fluorescence microscope Olympus BX61 (Olympus co., Tokyo, Japan).
SiRNA transfection
The p53 siRNA and control siRNA was purchased from biotechnology company (Shanghai GeneChem, Shanghai). Before transfection, MCF-7 cells which grew in completed growth medium were plated onto 6-well plates with a density of 1×106 cells/well. When cells grew to a confluence of 60%, transfection was performed by using Opti-MEM medium, lipofectamine 2000 and relevant siRNA according to the manufacturer's instruction. After 6 hours, the medium was replaced with completed growth medium. And cells were treated with 100μM cisplatin for 24 hours.
Statistical analysis
Comparisons were performed with a t‑test (unpaired or paired) by SPSS version 17.0 software (SPSS, Inc., Chicago, IL, USA). All P‑values presented were two‑tailed. P < 0.05 was considered to indicate a statistically significant difference.
Results
Cisplatin induced MCF-7 apoptosis in a concentration- and time-dependent manner
Human breast cancer cells, MCF-7, were treated with different concentration of cisplatin for 24h. The cells were measured by MTT assay, and it was showed that the percentage of survival was decreased markedly while the concentration of cisplatin increased (Figure 1A). And in the other test group, cells were exposed with the fixed concentration of cisplatin (100μM) but incubated for different times. MTT assay displayed that the number of death cells was increased with time elapsed (Figure 1B). This consequence was also confirmed in Hoechst 33342 staining test, the survival cells were continually disappeared from 6h to 24h.
Figure 1
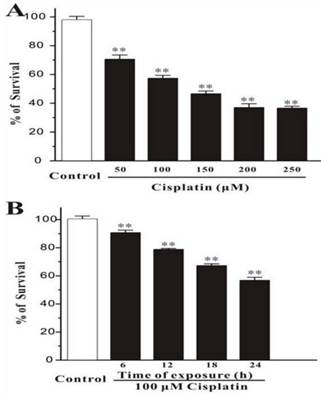
Cisplatin induces cell death in MCF-7 cells. (A) Cisplatin induces cell death in a concentration-dependent manner. MCF-7 cells were exposed to cisplatin at concentration as indicated for 24 h, and then cell viability was measured by MTT assay. (B) Cisplatin induces cell death in a time-dependent manner. MCF-7 cells were exposed to 100 μM cisplatin for different times as indicated, and then cell viability was measured by MTT assay. Data, expressed as percentage of control, were the mean ± SEM of three separate experiments, **p < 0.01 versus control group (unpaired t test).
Cisplatin up-regulates the expression of both AChE and p53
MCF-7 cells were treated with 100 μM cisplatin for different time, and expression of AChE and p53 proteins were detected by western blot. It was clearly found that both AChE and p53 proteins expressed increasingly in cisplatin-induced apoptotic cells caused by time increased (Figure 2). Using immunofluorescence to detect the expression of AChE proteins in cisplatin-induced apoptotic cells at different time, it was also significantly found that expression of AChE was elicited dramatically.
Silenced expression of AchE and p53 decrease cell apoptosis
All groups of MCF-7 treated with cisplatin (100μM) for 24h to stimulate cell apoptosis (Figure 3). One group was only used by cisplatin, and the other groups were added p53 siRNA, and control siRNA with cisplatin, respectively. Cells viability was determined by Hoechst 33342 staining, and statistically significance of data was analysised by Duncan's multiple range tests. It was observed that the number of survival cells in the p53 siRNA+cisplatin group is more than the cisplatin group by fluorescence microscope, whereas the cell condition in the control siRNA +cisplatin group was similar with the cisplatin group. Data was collected by counting the number of apoptotic cells in three different fields and the mean value was calculated. It was obviously showed that in control group only less than 5% of dead cells, while the percentage of apoptosis increased to 35% after added cisplatin. However, in the p53 siRNA+cisplatin group, the percentage of apoptotic cells was dramatically dropped to 15% and 20%, respectively. While in control siRNA and cisplatin group, the similar death proportion occurred with no significant difference.
Silencing of p53 by siRNA blocks cisplatin-induced AChE expression
MCF-7 cells were treated with100 μM cisplatin for 24 h in the absence or presence of p53 siRNA. The total proteins were analysed by western blot. It was clearly displayed that p53 and AChE were expressed increasingly when cells were treated with cisplatin compared with control. However, when presence of p53 siRNA with cisplatin, the expression of p53 was not only decreased, but AChE band also was weakened. And this situation was not occurred in the control siRNA+cisplatin group (Figure 4). Interestingly, comparison AChE isolated from nucleus and cytoplasm, the consequence showed that silencing of p53 by siRNA blocked the expression of AChE induced by cisplatin in the nuclear but not in cytoplasm (Figure 5).
Figure 2
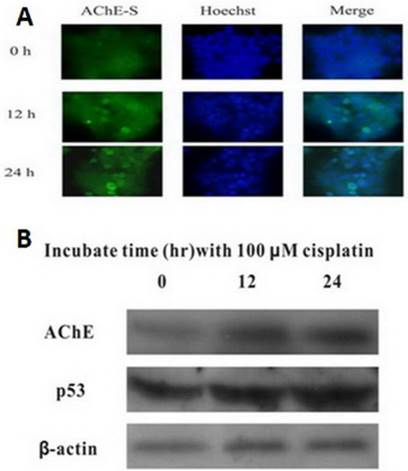
Cisplatin up-regulates the expression of both AChE and p53. MCF-7 cells were treated with 100 μM cisplatin for different time as indicated, and the total proteins were extracted for the western blot assay with the specific AChE, p53 and β-actin antibodies.
Figure 3
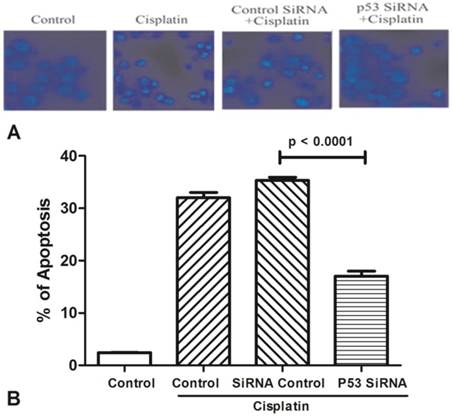
The expression of AChE is increased in the apoptosis cell induced by cisplatin. (A) MCF-7 cells were treated with 100 μM cisplatin for different time as indicated. And then AChE immunostaining and Hoechst staining were applied in the cells. (B) Inhibition of p53 or AChE expression attenuates the apoptosis induced by cisplatin. MCF-7 cells were treated with 100μM cisplatin for 24 h in the absence or presence of p53 or AChE siRNA. Then cells were conducted by Hoechst 33342 staining assay. The amount of apoptotic nuclei with condensed chromatin were counted from representative photomicrographs and were represented as a percentage of the total number of nuclei counted. Each treatment group was compared with the other groups using unpaired t test.
Figure 4
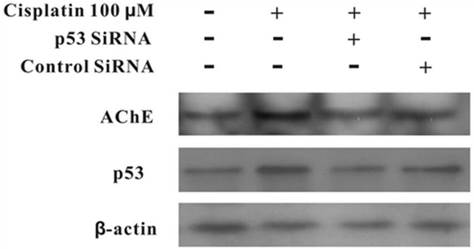
Silencing of p53 by siRNA blocks cisplatin-induced AChE expression. MCF-7 cells were treated with100 μM cisplatin for 24 h in the absence or presence of p53 siRNA. Then the total proteins were extracted for the western blot assay with the specific AChE, p53 and β-actin antibodies.
Figure 5
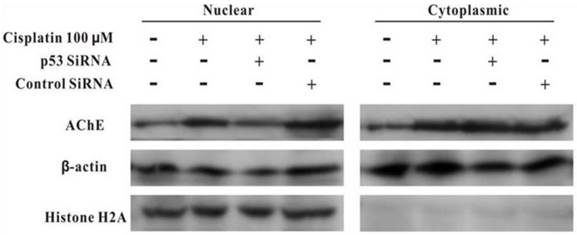
Silencing of p53 by siRNA blocks the expression of AChE induced by cisplatin in the nuclear but not in cytoplasm. MCF-7 cells were treated with100 μM cisplatin for 24 h in the absence or presence of p53 siRNA. Then the proteins either from nuclear or from cytoplasm were extracted for the western blot assay with the specific AChE, β-actin and Histone H2A antibodies.
Discussion
P53, as a regulating apoptotic protein, its major function has always been changed since 1979 when this protein was first reported. p53 could mediate cell cycle arrest and DNA damage, which is an important factor for apoptosis [17]. p53 could play an important regulator in cell apoptosis and tumorigenesis [10].
It implied that AChE play an important role in cell proliferation, differentiation, survival and tumorigenesis [18]. AChE is not deemed to be a simplex terminate synaptic transmission factor, but more unbeknown functions are being discovered and confirmed. Such as the role of AChE plays in the apoptotic process has been paid increasingly attention. The inhibition of AChE was able to decrease apoptosis and reduce the neurotoxin in model of Parkinson's disease [19]. Apoptosis can be prevented by blockage of AChE expression, and an increase of apoptosis has been found in retinal cells dues to transfection with AChE [20]. The interaction between apoptotic related factor-1 and cytochrome c is able to be inhibited through silencing of the siAChE, which to prevent apoptosis occurring [21]. In addition, AChE can be as a tumor suppressor gene, blocked caspase-9 activation to cause to decreased cell survival, nuclear condensation and adenosine diphosphate-ribose polymerase cleavage [22]. But the specific mechanism of AChE in apoptosis has not been recognized completely.
In this study, we confirmed that the expression of AChE was up-regulated in MCF-7 cells treated with cisplatin, what's more, blocked p53 was able to decrease cisplatin-induced AChE expression. It implied that AChE might be a downstream component part of p53 to induce the apoptosis in chemotherapy. Masha'our, R. S.et al. reported that retinoblastoma cells can escape apoptotic process when AChE protein was inhibited, which prove AChE as a pivotal factor in cancer apoptosis [23].
Although increasingly studies has focused on the function of AChE in apoptosis in various cellular types, few research studied about what influence on timorous cells would be exerted by AChE in chemotherapy. The previous results implied that the findings of p53-induced apoptosis and DNA-repair mechanisms are responsive to cisplatin-based chemotherapy in Testicular germ cell tumors [24].This study probably offered a new clue to derive another cancer therapeutic plan. However, further works need to expand to figure out the particular mechanism of AChE and even a new treatment which bases on the function of AChE in apoptosis is able to develop.
Acknowledgements
The present study was supported by the China Postdoctoral Science Foundation specific funded project (grant no. 201003380); the Natural Science Foundation of Ningbo (grant no. 2011A610057, 2011A610048); the Natural Science Foundation of China (grant no. 81372212); Jiangsu Provincial Special Program of Medical Science (grant no. BL2013012); the Health Talents Project for Jiangsu (grant nos. LJ201157; RC2011038).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Deng R, Li W, Guan Z, Zhou JM, Wang Y, Mei YP. et al. Acetylcholinesterase expression mediated by c-Jun-NH2-terminal kinase pathway during anticancer drug-induced apoptosis. Oncogene. 2006;25:7070-7 doi:10.1038/sj.onc.1209686
2. Ye W, Gong X, Xie J, Wu J, Zhang X, Ouyang Q. et al. AChE deficiency or inhibition decreases apoptosis and p53 expression and protects renal function after ischemia/reperfusion. Apoptosis: an international journal on programmed cell death. 2010;15:474-87 doi:10.1007/s10495-009-0438-3
3. Grisaru D, Sternfeld M, Eldor A, Glick D, Soreq H. Structural roles of acetylcholinesterase variants in biology and pathology. European journal of biochemistry / FEBS. 1999;264:672-86
4. Lionetto MG, Caricato R, Calisi A, Giordano ME, Schettino T. Acetylcholinesterase as a biomarker in environmental and occupational medicine: new insights and future perspectives. BioMed research international. 2013;2013:321213. doi:10.1155/2013/321213
5. Lawson AA, Barr RD. Acetylcholinesterase in red blood cells. American journal of hematology. 1987;26:101-12
6. Soreq H, Zakut H. Amplification of butyrylcholinesterase and acetylcholinesterase genes in normal and tumor tissues: putative relationship to organophosphorous poisoning. Pharmaceutical research. 1990;7:1-7
7. Ruiz-Espejo F, Cabezas-Herrera J, Illana J, Campoy FJ, Vidal CJ. Cholinesterase activity and acetylcholinesterase glycosylation are altered in human breast cancer. Breast cancer research and treatment. 2002;72:11-22
8. Ruiz-Espejo F, Cabezas-Herrera J, Illana J, Campoy FJ, Munoz-Delgado E, Vidal CJ. Breast cancer metastasis alters acetylcholinesterase activity and the composition of enzyme forms in axillary lymph nodes. Breast cancer research and treatment. 2003;80:105-14 doi:10.1023/A:1024461108704
9. Soreq H, Lapidot-Lifson Y, Zakut H. A role for cholinesterases in tumorigenesis? Cancer cells. 1991;3:511-6
10. Levine AJ, Hu W, Feng Z. The P53 pathway: what questions remain to be explored? Cell death and differentiation. 2006;13:1027-36 doi:10.1038/sj.cdd.4401910
11. Levine AJ, Finlay CA, Hinds PW. P53 is a tumor suppressor gene. Cell. 2004;116:S67-9
12. Shen Y, White E. p53-dependent apoptosis pathways. Advances in cancer research. 2001;82:55-84
13. Schuler M, Green DR. Mechanisms of p53-dependent apoptosis. Biochemical Society transactions. 2001;29:684-8
14. Jin QH, He HY, Shi YF, Lu H, Zhang XJ. Overexpression of acetylcholinesterase inhibited cell proliferation and promoted apoptosis in NRK cells. Acta pharmacologica Sinica. 2004;25:1013-21
15. Pegan K, Matkovic U, Mars T, Mis K, Pirkmajer S, Brecelj J. et al. Acetylcholinesterase is involved in apoptosis in the precursors of human muscle regeneration. Chemico-biological interactions. 2010;187:96-100 doi:10.1016/j.cbi.2010.03.034
16. Zhao Y, Wang X, Wang T, Hu X, Hui X, Yan M. et al. Acetylcholinesterase, a key prognostic predictor for hepatocellular carcinoma, suppresses cell growth and induces chemosensitization. Hepatology. 2011;53:493-503 doi:10.1002/hep.24079
17. Liebermann DA, Hoffman B, Vesely D. p53 induced growth arrest versus apoptosis and its modulation by survival cytokines. Cell cycle. 2007;6:166-70
18. Jiang H, Zhang XJ. Acetylcholinesterase and apoptosis. A novel perspective for an old enzyme. The FEBS journal. 2008;275:612-7 doi:10.1111/j.1742-4658.2007.06236.x
19. Zhang X, Lu L, Liu S, Ye W, Wu J, Zhang X. Acetylcholinesterase deficiency decreases apoptosis in dopaminergic neurons in the neurotoxin model of Parkinson's disease. The international journal of biochemistry & cell biology. 2013;45:265-72 doi:10.1016/j.biocel.2012.11.015
20. Zhang XJ, Yang L, Zhao Q, Caen JP, He HY, Jin QH. et al. Induction of acetylcholinesterase expression during apoptosis in various cell types. Cell death and differentiation. 2002;9:790-800 doi:10.1038/sj.cdd.4401034
21. Park SE, Kim ND, Yoo YH. Acetylcholinesterase plays a pivotal role in apoptosome formation. Cancer research. 2004;64:2652-5
22. Zhang XJ, Greenberg DS. Acetylcholinesterase involvement in apoptosis. Frontiers in molecular neuroscience. 2012;5:40. doi:10.3389/fnmol.2012.00040
23. Masha'our RS, Heinrich R, Garzozi HJ, Perlman I. Acetylcholinesterase (AChE) is an important link in the apoptotic pathway induced by hyperglycemia in Y79 retinoblastoma cell line. Frontiers in molecular neuroscience. 2012;5:69. doi:10.3389/fnmol.2012.00069
24. Cavallo F, Feldman DR, Barchi M. Revisiting DNA damage repair, p53-mediated apoptosis and cisplatin sensitivity in germ cell tumors. The International journal of developmental biology. 2013;57:273-80 doi:10.1387/ijdb.130135mb
Author contact
![]() Corresponding author: Tianbao Zhou, The Hepatobiliary Surgery Centre, The Ningbo No. 2 Hospital, Ningbo 315010, China. Phone: +86-519-86867281; Fax: +86-519-86867281; E-mail: zhangchangsongedu.cn.
Corresponding author: Tianbao Zhou, The Hepatobiliary Surgery Centre, The Ningbo No. 2 Hospital, Ningbo 315010, China. Phone: +86-519-86867281; Fax: +86-519-86867281; E-mail: zhangchangsongedu.cn.
Citation styles
APA
Ye, X., Zhang, C., Chen, Y., Zhou, T. (2015). Upregulation of Acetylcholinesterase Mediated by p53 Contributes to Cisplatin-Induced Apoptosis in Human Breast Cancer Cell. Journal of Cancer, 6(1), 48-53. https://doi.org/10.7150/jca.10521.
ACS
Ye, X.; Zhang, C.; Chen, Y.; Zhou, T. Upregulation of Acetylcholinesterase Mediated by p53 Contributes to Cisplatin-Induced Apoptosis in Human Breast Cancer Cell. J. Cancer 2015, 6 (1), 48-53. DOI: 10.7150/jca.10521.
NLM
Ye X, Zhang C, Chen Y, Zhou T. Upregulation of Acetylcholinesterase Mediated by p53 Contributes to Cisplatin-Induced Apoptosis in Human Breast Cancer Cell. J Cancer 2015; 6(1):48-53. doi:10.7150/jca.10521. https://www.jcancer.org/v06p0048.htm
CSE
Ye X, Zhang C, Chen Y, Zhou T. 2015. Upregulation of Acetylcholinesterase Mediated by p53 Contributes to Cisplatin-Induced Apoptosis in Human Breast Cancer Cell. J Cancer. 6(1):48-53.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) License. See http://ivyspring.com/terms for full terms and conditions.