Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2014; 5(8):655-662. doi:10.7150/jca.6554 This issue Cite
Research Paper
Down Regulation of FOXO1 Promotes Cell Proliferation in Cervical Cancer
Shyam Babu Prasad1, Suresh Singh Yadav1, Mitali Das1, H. B. Govardhan2, Lakshmi Kant Pandey3, Sunita Singh4, Satyajit Pradhan2, Gopeshwar Narayan1 ![]()
1. Cancer Genetics Laboratory, Department of Molecular and Human Genetics, Banaras Hindu University, Varanasi-221 005, India;
2. Department of Radiotherapy and Radiation Medicine, Banaras Hindu University, Varanasi-221 005, India;
3. Department of Obstetrics and Gynaecology, Banaras Hindu University, Varanasi-221 005, India;
4. Department of Zoology, Mahila Mahavidyalaya; Banaras Hindu University, Varanasi-221 005, India.
Received 2013-4-24; Accepted 2013-7-9; Published 2014-8-22
Abstract
The Forkhead transcription factor FOXO1, an important downstream target of phosphatidylinositol-3-kinase (PI3K)/AKT signaling pathway, regulates cellular homeostasis by maintaining cell proliferation, apoptosis and viability in normal cells. Though, the function and regulation of FOXO1 is well documented in many cancers, the molecular mechanism of its regulation in cervical cancer is largely unknown. In the present study we have investigated the role of PI3K inhibition on FOXO1 regulation. Expression profiling of primary tumors and cell lines show over expression of PIK3CA and AKT1; and down regulation of FOXO1. Lack of FOXO1 promoter methylation and inability of hypomethylating drug 5-Aza-2'-deoxycytidine and HDAC inhibitor trichostatin A to reactivate FOXO1 expression suggest that loss of FOXO1 expression is due to mechanisms other than promoter methylation/acetylation. Inhibition of PI3K by LY294002 decreased the level of p-AKT1 and activated FOXO1 transcription factor. We demonstrate that activation of FOXO1 induces apoptosis, cell proliferation arrest, and decreased cell viability in cervical cancer cell lines. Our data suggest that frequent down regulation of FOXO1 and its functional inactivation may be due to post-translational modifications in cervical cancer. Together, these observations suggest that activation of FOXO1 and its nuclear sequestration is critical in the regulation of cell proliferation, cell viability and apoptosis in cervical cancer. Hence, PI3K/AKT pathway may be a potential molecular target for cervical cancer therapy.
Keywords: Cervical cancer, PI3K/AKT, FOXO1, LY294002, Apoptosis.
Introduction
The phosphatidylinositol-3-kinase (PI3K) signaling pathway is a crucial regulator of many normal cellular processes, such as cell growth, proliferation, motility, survival, and apoptosis. Deregulation of several components of this pathway, including, PIK3CA, AKT and PTEN have been well documented in a wide range of human cancers [1-3]. PIK3CA, a key element of the PI3K/AKT pathway, is located at chromosomal region 3q26.3, and encodes the 110kDa catalytic subunit of class IA (PI3K). PI3K plays a role in phosphorylation of FOXO proteins via activation of its downstream kinase AKT1 [4-6]. FOXO family includes three functionally related members of forkhead transcription factors FOXO1a/FKHR, FOXO3a/FKHRL1, and FOXO4/AFX [7-8]. The FOXO1 transcriptional activity is controlled by its post-translational modification at two or three conserved residues (Thr24, Ser256, and Ser319) resulting in its nuclear exclusion [9-11]. The phosphorylation of FOXO1 results in the impairment of its DNA binding ability and increased binding affinity for 14-3-3 protein [4-6]. Newly formed 14-3-3-FOXO complex is then exported from the nucleus thereby inhibiting FOXO1 dependent transcription [12]. Inhibition of the PI3K pathway leads to dephosphorylation and nuclear translocation of active FKHRL1, FKHR, and AFX leading to cell growth arrest and apoptosis [13]. FOXO1 regulates cell cycle regulatory proteins such as cyclin-dependent kinase inhibitor (CKI/p27kip1) [14-16], and apoptosis mediators Bim, Fas ligand and Bcl-6 [17, 1, 18]. FOXO1 is down regulated in several cancers including endometrial carcinoma [19]. Over expression of AKT1, which modulates FOXO1 activity, is frequently observed in pancreatic cancer, indicating a potential role of FOXO1 in regulation of cell cycle, proliferation and apoptosis during tumorigenesis [20-22]. Since FOXO1 activity is involved in the regulation of important cellular decision and maintenance of normal cellular homeostasis, loss of its activity either by down regulation or by post-transcriptional/post-translational modifications leads to cancer development [9, 19, 23]. Therefore, understanding of the molecular mechanisms of the regulation of FOXO1 transcription factors may be useful in identification of molecular therapeutic targets in cervical cancer (CC). Also, the ability of FOXO1 to induce cell growth arrest, DNA repair and apoptosis makes it an attractive candidate as a tumor suppressor [24].
In the present study, we hypothesized that down regulation of FOXO1 and/or its inactivation by post-translational modification may have a critical role in cervical cancer progression. Correlation of FOXO1 expression with clinicopathological parameters may provide a clue for early diagnosis.
Materials and Methods
Sample collection and maintenance of CC cell lines
Normal and primary tumor samples were collected from Institute of Medical Sciences (IMS), Banaras Hindu University (BHU), Varanasi as per approved protocol by the institutional ethical committee of the IMS, BHU after patient's written informed consent. Eight cervical cancer cell lines (HeLa, SiHa, ME-180, CaSki, C-33A, C-4I, SW756 and MS751) obtained from American Type Cell Culture (ATCC, Mansas, VA) that have been extensively characterized [25-28] were kind gift from Dr. VVVS Murty (Columbia University, New York, USA). The cell lines were maintained in ATCC recommended media and 10% heat-inactivated fetal bovine serum (FBS) at 37°C in a humidified atmosphere with 5% CO2.
Reverse Transcriptase-PCR and Real Time-PCR
Biopsy samples and cell lines were lysed using TRI Reagent (Sigma, USA) and total RNA was extracted as per manufacturer's protocol. The first strand cDNA was synthesized using commercially available high capacity cDNA reverse transcription kit (ABI, USA) as per manufacturer's protocol. PCR of cDNA was done in triplicate using gene specific primers (Supplementary Material: Table S1). Samples were then electrophoresed on a 1.8% agarose gel and visualized using ethidium bromide staining. Real-time PCR (qRT-PCR) was performed with 2X SYBR Green PCR Master Mix (ABI, USA) according to the manufacturer's protocol. The threshold cycle (CT) values were determined using ABI 7500 sequence detection system and the data was analyzed with CT values of β-actin as reference (endogenous control).
Bisulfite conversion of DNA for Methyl specific PCR (MSP)
The proximal FOXO1 promoter region was analyzed for the presence of CpG islands using the CpG island searcher (www.cpgislands.com). CpG-rich region of FOXO1 promoter, were considered as a stretch of DNA that has both a >50% GC content and an observed over expected frequency of CpG dinucleotides of >0.6 [29]. Phenol/Chloroform extracted genomic DNA from normal biopsy, primary tumors and CC cell lines was used for bisulfite conversion using EpiTech bisulfite conversion kit (Qiagen GmbH, Germany) by manufacturer's protocol. The proximal FOXO1 gene promoter primers were designed by Methyl Express software V1.0 (ABI, UK) for the region harboring CpG-rich islands covering -1758 to +242 bp including the transcription start site (TSS) (Supplementary Material: Fig. S1). MSP was performed using 2x PCR master mix (Fermentas, USA) with three sets of primer sequences (Supplementary Material: Table S1) for -985 to -645 bp (MSP1); -645 to -285bp (MSP2) and -225 to +242bp (MSP3) in triplicate. Placental DNA treated in vitro with SssI methyltransferase (NEB, Beverly, MA) and lymphocyte DNA were bisulfite converted and used as methylated and unmethylated controls respectively. PCR was performed using standard conditions for 30-35 cycles with annealing temperatures varying between 58ºC and 63ºC. For qualitative assessment of methylation status of FOXO1, the PCR products were electrophoresed on a 1.8% agarose gel and visualized using ethidium bromide staining.
Drug treatments
The cell lines HeLa, SiHa, ME180 and SW756 were treated with 5-Aza-2'-deoxycytidine (AZA) (Sigma, USA) and HDAC inhibitor Trichostatin A (TSA) (Sigma, USA) as described previously in [25] to reactivate FOXO1 transcription. For cell proliferation, cell viability and apoptosis, cells were initially grown in 96-well plate (1x104 cells/well) and 6-well plate (1x105 cells/well) for 24hr, followed by treatment with 10µM and 25 µM of LY294002 for 48hr.
Western blot analysis
Total cell lysates were obtained by lysing biopsy samples and cell lines with RIPA buffer containing 100mM NaCl, 50mM Tris-Cl (pH 7.4), 2mM EGTA, 1mM EDTA, 1mM DTT, 1mM PMSF 1% NP-40, 0.1% SDS plus protease inhibitor cocktail (Sigma, USA) on ice. The whole cell proteins were isolated and concentration was measured using Bradford assay. Equal amounts of protein (50 and 100μg) were separated using 12% SDS-PAGE and transferred to PVDF membrane (Millipore Corporation, Billerica, MA, USA). Membranes were blocked with 5% nonfat milk in TBST at room temperature and incubated with anti-FOXO1 primary antibody (Abcam, Cambridge, MA, USA), anti-p-FOXO1 (Ser256) from (Cell Signaling, Beverly, MA, USA), anti-AKT1 (Imgenex, India), anti-p-AKT1 (Thr308) (Santa Cruz Biotechnology, USA) in 1% nonfat milk in TBST overnight at 4ºC. After the incubation, membranes were washed three times with TBST and incubated in secondary ALP-conjugated goat anti-rabbit IgG or goat anti-mouse IgG (Bangalore Genie, India) in 1% nonfat milk in TBST. The membranes were developed with the NBT/BCIP solution (Amresco, USA) after washing. The membranes were stripped and re-probed with anti-β-actin antibody (Pierce, USA) as a loading control. All the experiments were repeated thrice.
Immunofluorescent staining
Immunofluorescence was performed on cell lines grown on cover slips in standard culture condition. The cell lines were treated with different concentrations of LY294002 for 48hr. After treatment the cells were washed in 1x PBS and fixed with 4% paraformaldehyde in 1x PBS for 15 min at room temperature. Cells were then permeabilized by incubating with 0.1% Triton X-100 in PBS for 10 min at room temperature. After washing, the cells were incubated in blocking solution (1% BSA) for 30 min at RT. The cell lines were incubated with anti-p-AKT1 (Thr308) (Santa Cruz Biotechnology, USA), anti-human p-FOXO1 (Ser256) (Cell Signaling, Beverly, MA, USA) overnight at 4˚C. After washing, cells were incubated with Cy5 and FITC-labeled goat anti-rabbit IgG secondary antibodies for 2hr at 4˚C, and nuclei were then counter-stained with DAPI (Sigma, USA). Cells were mounted with DABCO (Sigma, USA) mounting medium and visualized using a fluorescence microscope (Nikon Eclipse 80i). Each experiment was performed in triplicate.
Cell proliferation, viability assay and analysis of apoptosis by flow cytometry
Cell proliferation was measured using the Cell Proliferation Assay Kit (Millipore, USA), according to the manufacturer's protocol in triplicate. Briefly, 10µl of WST-1/ECS solution was added to each well, the samples were incubated for 4hr at standard culture conditions and absorbance was measured at 450nm. Cell viability were counted by automated cell counter (TC-10 Biorad, USA) for treated and untreated cell lines by standard trypan blue exclusion method.
For apoptosis assay cells were exposed to different concentrations of LY292002 for 48hr at standard culture conditions. For cell death assay, cells were harvested and washed twice in phosphate buffered saline and stained with AnnexinV-AlexaFlor488 and propidium iodide (Vibrant apoptosis detection kit, Invitrogen, USA) according to the manufacturer's protocol. Each sample was analyzed by fluorescence activated cell sorter (FACS) (BD, San Jose, CA, USA).
Statistical analysis
Data was expressed as mean ± SEM; for comparisons between more than 2 groups, ANOVA followed by the Bonferroni multiple comparison post hoc tests; and for comparisons between 2 groups, unpaired 2-tailed Student's t-test were applied using GraphPad Prism5 software (La Jolla, USA). The p-values less than 0.05 were considered as statistically significant.
Results
Frequent down regulation of FOXO1 and up regulation of PI3K-AKT1 in cervical cancer cell lines and primary tumors
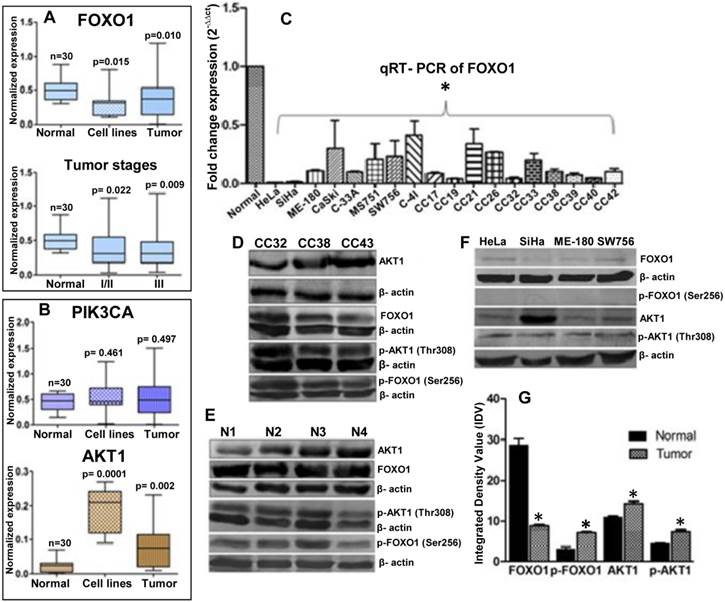
Semi-quantitative RT-PCR were performed for expression profiling of PIK3CA, AKT1 and FOXO1 in eight cervical cancer cell lines (HeLa, SiHa, ME-180, CaSki, C33A, MS751, C-4I and SW756), 70 primary tumor biopsy and 30 normal cervical tissue samples. Results show that FOXO1 is frequently down regulated (41%) (Fig. 1A) where as PIK3CA (25%) and AKT1 (45%) are frequently up regulated (Fig. 1B), in primary tumors and cell lines. The quantitative RT-PCR shows more than tenfold down regulation of FOXO1 (Fig. 1C). Similarly, our immunoblotting data also shows up regulation of AKT1 and down regulation of FOXO1 (Fig. 1D, 1E 1F and 1G). FOXO1 was significantly down regulated irrespective of the tumor stages: stages I/II (p=0.002) and III (p=0.009) (Fig. 1A). While p-AKT1 (Thr308) is up regulated in primary tumors and cell lines, p-FOXO1 (Ser256) is elevated in primary tumors (Fig. 1D, 1G) but undetectable in the cell lines (Fig. 1F). Moreover, while native AKT1 is detectable in all the cell lines, FOXO1 show very low expression in cell lines.
Expression profiles of the genes in normal, tumor biopsy samples and cell lines: (A) mRNA expression levels of FOXO1 in CC cell lines (n=8); and normal (n=30), primary tumor Stage I/II (n=23) and III (n=30) biopsy samples. (B) Expression profile of PIK3CA and AKT1 in eight CC cell lines and different primary tumors. (C) FOXO1 mRNA was measured in eight cell lines and primary tumor biopsy using qRT-PCR. All samples were normalized with β-actin expression (endogenous control). Note the down-regulation of FOXO1 compared to normal biopsy (Bars represent mean ± SEM; *p<0.05 vs control; ANOVA-test was employed). (D, E, F) Whole cell lysate of biopsy samples (CC), normal (N) and cell lines were used for western blot analysis by using full length anti-FOXO1, anti-AKT1 and anti-p-FOXO1 (Ser256), anti-p-AKT1 (Thr308) antibodies. (G) The band intensities were estimated by densitometry as the integrated density value (IDV) for each protein band and normalized by the IDV of β-actin. The anti-β-actin antibody was used as protein loading control. *P <0.05 was considered statistically significant (unpaired 2-tailed Student's t-test).
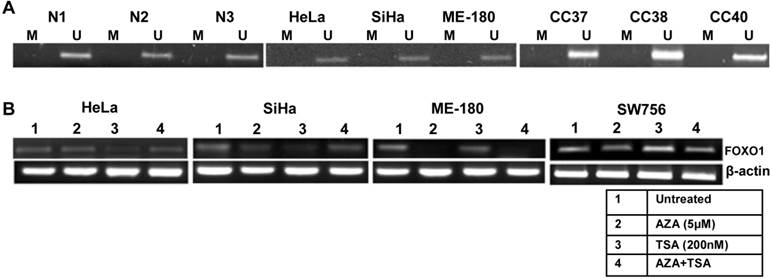
Loss of FOXO1 expression is not due to epigenetic silencing
To explain the mechanism of the down regulation of the FOXO1 expression, we checked its promoter methylation status. Our MSP results for all the three regions (Supplementary Material: Fig. S1) show that the promoter of FOXO1 is unmethylated in cell lines and primary tumors similar to the normal samples (Fig. 2A). Treatments with demethylating agent 5'-Aza-2-deoxycytidine (AZA) and/or HDAC inhibitor trichostatin A (TSA) did not reactivate the FOXO1 expression (Fig. 2B), indicating that the inactivation of the FOXO1 is not due to promoter hypermethylation or histone acetylation.
Methylation pattern of the proximal promoter region and reactivation of FOXO1 in cervical cancer: (A) Methyl specific-PCR (MSP) analysis showing unmethylated proximal FOXO1 promoter (2kb of CpG island) including transcription start site (TSS) in normal (N), primary tumor biopsy (CC) and cell lines. Promoter hypermethylation/unmethylation was considered positive when present in at least one of the regions in two independent experiments. (B) 5'-Aza-2-deoxycytidine (AZA) and trichostatin A (TSA) treated HeLa, SiHa, ME-180 and SW756 cell lines. Note that the RT-PCR does not show reactivation of the FOXO1 expression.
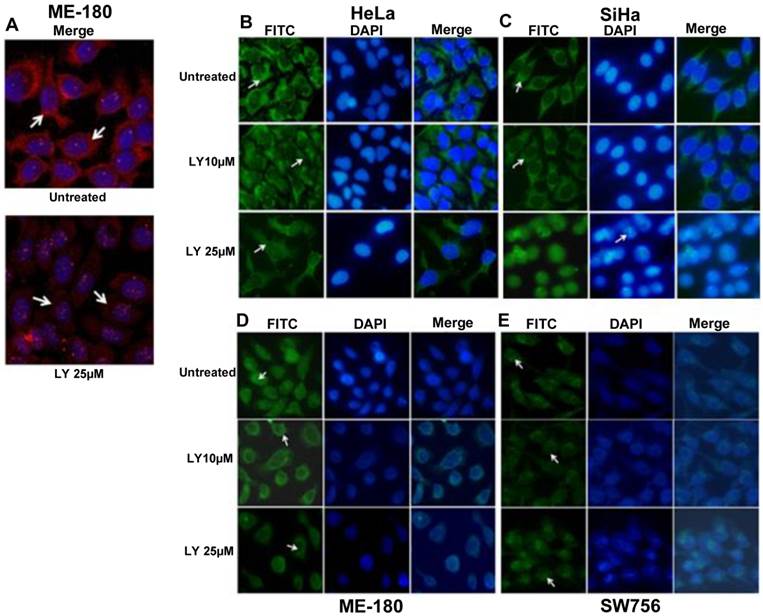
Inhibition of PI3K activates FOXO1 and sequesters it to the nucleus
PI3K/AKT pathway has been shown to regulate the phosphorylation of FOXO proteins [30] leading to its functional inactivation and cytoplasmic sequestration. To determine whether the change of FOXO1 localization in CC cell lines was due to AKT1 activation, we used PI3K inhibitor LY294002 (LY25µM; 48hr) to suppress p-AKT1 (Thr308) activation. Immunofluorescence data show remarkably reduced level of p-AKT1 (Thr308) in ME-180 compared to untreated (Fig. 3A). The p-FOXO1 (Ser256) is localized in the cytoplasm in both untreated and 10μM LY294002 (LY10µM) treated SiHa and HeLa cell lines, whereas following 25μM LY294002 treated SiHa cells show reduced cytoplasmic p-FOXO1 (Fig. 3C). Morphological changes in nuclear architecture, like nuclear blebbing and chromatin condensation, characteristic feature of early apoptotic cells (Fig. 3C), was also observed. Treated HeLa cells (LY25µM; 48hr) show weak dispersed staining for p-FOXO1 protein throughout the cytoplasm (Fig. 3B). However, the perinuclear and nuclear expression of p-FOXO1 (Ser256) were seen in ME-180 and SW756 (Fig. 3D, 3E) instead of cytoplasmic localization.
Fluorescence microscopy for the p-FOXO1 (Ser256) localization in LY294002 treated cervical cancer cell lines: (A) ME-180 cells were treated with PI3K inhibitor LY294002 (LY25μM) for 48hr. Immunofluorescence result show reduced cytoplasmic level (indicated by arrow head) of anti-p-AKT1 (Thr308). (B, C) p-FOXO1 (Ser256) localization in the cytoplasm (indicated by arrow head) in both SiHa and HeLa cell lines in untreated and treated cells with LY294002 (LY10μM) drug. LY294002 (LY25μM) treated SiHa cells show only nuclear localization of p-FOXO1 and also nuclear blebbing and chromatin condensation (indicated arrow head) in DAPI panel. HeLa cells show absence of cytoplasmic p-FOXO1 (indicated by arrow head) after LY294002 (LY25μM) treatment for 48hr. (D, E) Perinuclear and nuclear localization (indicated by arrow head) of p-FOXO1 (Ser256) in ME-180 and SW756 cells treated with LY294002 for 48hr.
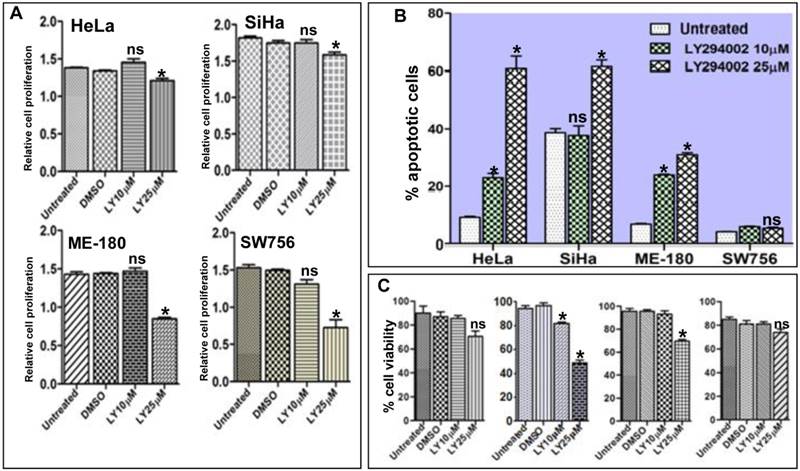
Activation of FOXO1 attenuates cells proliferation and viability; and induces apoptosis in CC cell lines
It is well documented that inhibition of the PI3K/AKT pathway results in activation of FOXO1 and restores its transcriptional activity which acts as transcription factor for many genes involved in apoptotic pathway [31]. We hypothesized that reactivation of FOXO1 should attenuate cell proliferation, viability and induce apoptosis in CC cell lines. Proliferation assay results show significant inhibition of cell proliferation (Fig. 4A). The Flow cytometry assay confirmed that the fraction of apoptotic cells was considerably higher and statistically significant in SiHa, HeLa and ME-180 than SW756 cells after 48hr of LY294002 treatment (Fig. 4B and Supplementary Material: Fig. S2). Loss of cell viability was much more pronounced in SiHa than HeLa, ME-180 and SW756 when exposed to LY294002 (25µM; 48hr) (Fig. 4C), indicating reactivation of FOXO1.
Activation of FOXO1 attenuates cell proliferation, viability and induces apoptosis in cell lines: (A) HeLa, SiHa, ME-180 and SW756 cells show the relative inhibition of cell proliferation (absorbance measured at 460nm) after the treatment with 10μM and 25μM of LY294002 or without LY294002 (untreated control) and DMSO (vehicle control) for 48hr. (B) Percentage of apoptotic cells after LY294002 treatment. (C) Percentage cell viability after LY294002 treatment. SiHa shows frequent loss of viable cells at both the concentrations (10μM and 25μM) than other three cell lines. Bars represent mean ± SEM: *p<0.05 vs. control.
Discussion
PI3K/AKT signaling pathway has been associated with cancer progression and has critical role in tumor development. FOXO1 is an important downstream effecter of this pathway; in the present study, we have attempted to investigate the consequences of the inhibition of PI3K with LY294002 on FOXO1 regulation and cellular endpoints.
Frequent over expression of the PIK3CA and AKT1 in primary tumors and CC cell lines in our study is consistent with previously reported 3q26.3 amplification with increased copy number of PIK3CA [32] and over representation of AKT1 at 14q32.33 [33] suggesting deregulation of the PI3K/AKT signaling pathway in cervical cancer. Interestingly, our data also show significant down regulation of FOXO1 expression in primary tumors and CC cell lines, similar to previous studies in non-small cell lung cancer [34]; and endometroid endometrial carcinoma (EEC) and non-endometrioid cancers [35]. Lack of promoter methylation of FOXO1 revealed by MSP in primary tumors and cell lines and inability of demethylating agent AZA and HDAC inhibitor TSA to reactivate FOXO1 expression in cell lines, suggest that methylation/acetylation may not be a cause for loss of FOXO1 expression in cervical cancer. The loss of FOXO1 expression may correlate with high turnover of mRNA transcript as previously shown [19] and miRNA mediated repression of FOXO1 expression as reported in endometrial and breast cancer cells [23, 36]. In addition to miRNA, other mechanism(s) may contribute to loss of FOXO1 expression in cervical cancer. The western blot analyses, show down regulated expression of FOXO1 protein and undetectable p-FOXO1 (Ser256) level in CC cell lines. The presence of active p-AKT1 (Thr308) suggests post-translational modification/degradation of FOXO1 in CC cell lines. The previous reports of post-translational modification and proteasomal degradation in endometrial cancer [37] also support our data. Reduced cytoplasmic p-FOXO1 in cytoplasm indicating its nuclear translocation at 25µM of LY294002 treated SiHa and HeLa cells, where FOXO1 functions as a transcriptional activator for death genes [38], induces apoptosis (Fig. 3C). The perinuclear/nuclear expression of p-FOXO1 (Ser256) in ME-180 and SW756 cell lines, suggest that either some other proteins such as GSK may play important role in FOXO1 phosphorylation [39] or mutation in FOXO1 gene may prevent its cytoplasmic translocation. We further show that inhibition of PI3K by LY294002 decreases cell proliferation and induces apoptosis in cervical cancer cells similar to induced apoptosis in prostate cancer cells [31] and in H23 lung adenocarcinoma cells [40] by resveratrol and LY294002 respectively. The significant inhibition of cell proliferation in HeLa, SiHa, ME-180 and SW756 may be due to the reduced phosphorylation of FOXO1. Statistically significant high number of apoptotic cells in SiHa, HeLa and ME-180 after 25µM of LY294002 treatment further suggests activation of FOXO1. Similarly, loss of cell viability on exposure to LY294002 supports the reduced cell proliferation and induction of apoptosis. Similar to apoptosis, more pronounced loss of cell viability in SiHa at 25µM of LY294002 further supports that inhibition of PI3K activates FOXO1 transcription factor. Based on our current knowledge and available data to date in the cervical cancer, this is the first report of down regulation of FOXO1 and its PI3K/AKT signaling pathway mediated post-translational regulation in cervical cancer.
In conclusion, the frequent over expression of PIK3CA and AKT1 suggests deregulation of PI3K/AKT signaling pathway. Down regulation of FOXO1 and lack of promoter methylation demonstrate that FOXO1 activity may be regulated by post-translational modification(s). Inhibition of PI3K induces activation of FOXO1 leading to cell proliferation arrest, induction of apoptosis and reduced cell viability in CC cell lines. Hence, PI3K/AKT pathway is critical in regulation of FOXO1 activity. Therefore, detailed understanding of the mechanism of PI3K/AKT/FOXO1 pathway regulation may contribute in identification of novel molecular therapeutic target(s) of cervical cancer.
Supplementary Material
Figures S1-S2, Table S1.
Acknowledgements
Department of Biotechnology (DBT), Ministry of Science and Technology, India for the financial assistance (BT/PR9246/Med/30/17/2007) to GN. Indian Council of Medical Research (ICMR) for providing Senior Research fellowship to SBP. Authors are thankful to Dr. VVVS Murty (Columbia University, New York, USA) for kind gift of cervical cancer cell lines.
Conflict of Interest
The authors declare that they have no competing conflict of interest.
References
1. Vivanco I, Sawyers CL. The phosphatidylinositol-3-kinase AKT pathway in human cancer. Nat Rev Cancer. 2002;2:489-1
2. Vogelstein B, Kinzler KW. Cancer genes and the pathways they control. Nat Med. 2004;10:789-9
3. Cully M, You H, Levine AJ, Mak TW. Beyond PTEN mutations: the PI3K pathway as an integrator of multiple inputs during tumorigenesis. Nat Rev Cancer. 2006;6:184-92
4. Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 1999;96:857-8
5. Van Der Heide LP, Hoekman MF, Smidt MP. The ins and outs of FoxO shuttling: mechanisms of FoxO translocation and transcriptional regulation. Biochem J. 2004;380:297-9
6. Guo S, Rena G, Cichy S, He X, Cohen P, Unterman T. Phosphorylation of serine-256 by protein kinase B disrupts transactivation by FKHR and mediates effects of insulin on insulin-like growth factor-binding protein-1 promoter activity through a conserved insulin response sequence. J Biol Chem. 1999;274:17184-92
7. Galili N, Davis RJ, Fredericks WJ, Mukhopadhyay S, Rauscher FJ, Emanuel BS, Rovera G, Barr FG. Fusion of a fork head domain gene to PAX3 in the solid tumour alveolar rhabdomyosarcoma. Nat Genet. 1993;5:230-35
8. Borkhardt A, Repp R, Haas OA, Leis T, Harbott J, Kreuder J, Hammermann J, Henn T, Lampert F. Cloning and characterization of AFX, the gene that fuses to MLL in acute leukemias with a t(X;11)(q13;q23). Oncogene. 1997;14:195-2
9. Fukunaga K, Ishigami T, Kawano T. Transcriptional regulation of neuronal genes and its effect on neural functions: expression and function of forkhead transcription factors in neurons. J Pharmacol Sci. 2005;98:205-11
10. Rena G, Guo SD, Cichy SC, Unterman TG, Cohen P. Phosphorylation of the transcription factor forkhead family member FKHR by protein kinase B. J Biol Chem. 1999;274:17179-83
11. Zhang X, Gan L, Pan H, Guo S, He X, Olson ST, Mesecar A, Adam S, Unterman TG. Phosphorylation of Serine 256 Suppresses Transactivation by FKHR (FOXO1) by Multiple Mechanisms. J Biol Chem. 2002;277:45276-84
12. Medema RH, Kops GJ, Bos JL, Burgering BM. AFX-like Forkhead transcription factors mediate cell-cycle regulation by Ras and PKB through p27kip1. Nature. 2000;404:782-7
13. Nakamura N, Ramaswamy S, Vazquez F, Signoretti S, Loda M, Sellers WR. Forkhead transcription factors are critical effectors of cell death and cell cycle arrest downstream of PTEN. Mol Cell Biol. 2000;20:8969-82
14. Dijkers PF, Medema RH, Pals C, Banerji L, Thomas NS, Lam EW, Burgering BM, Raaijmakers JA, Lammers JW, Koenderman L, Coffer PJ. Forkhead transcription factor FKHR-L1 modulates cytokine-dependent transcriptional regulation of p27 (KIP1). Mol Cell Biol. 2000;20:9138-48
15. Cappellini A, Tabellini G, Zweyer M, Bortul R, Tazzari PL, Billi AM, Fala F, Cocco L, Martelli AM. The phosphoinositide 3-kinase/Akt pathway regulates cell cycle progression of HL60 human leukemia cells through cytoplasmic relocalization of the cyclin-dependent kinase inhibitor p27 (Kip1) and control of cyclin D1 expression. Leukemia. 2003;17:2157-67
16. Burgering BM, Kops GJ. Cell cycle and death control: long live forkheads. Trends Biochem. Sci. 2002;27:352-60
17. Dijkers PF, Medema RH, Lammers JW, Koenderman L, Coffer PJ. Expression of the pro-apoptotic Bcl-2 family member Bim is regulated by the forkhead transcription factor FKHR-L1. Curr Biol. 2000;10:1201-4
18. Tang TT, Dowbenko D, Jackson A, Toney L, Lewin DA, Dent AL, Lasky LA. The forkhead transcription factor AFX activates apoptosis by induction of the BCL-6 transcriptional repressor. J Biol Chem. 2002;277:14255-65
19. Goto T, Takano M, Albergaria A, Briese J, Pomeranz KM, Cloke B, Fusi L, Feroze-Zaidi F, Maywald N, SAjin M, Dina RE, Ishihara O, Takeda S, Lam EW, Bamberger AM, Ghaem-Maghami S, Brosens JJ. Mechanism and functional consequences of loss of FOXO1 expression in endometrioid endometrial cancer cells. Oncogene. 2008;27:9-19
20. Altomare DA, Tanno S, De Rienzo A, Klein-Szanto AJ, Tanno S, Skele KL, Hoffman JP, Testa JR. Frequent activation of AKT2 kinase in human pancreatic carcinomas. J Cell Biochem. 2003;88:470-76
21. Perugini RA, McDade TP, Vittimberga FJ Jr, Callery MP. Pancreatic cancer cell proliferation is phosphatidylinositol 3-kinase dependent. J Surg Res. 2000;90:39-44
22. Schlieman MG, Fahy BN, Ramsamooj R, Beckett L, Bold RJ. Incidence, mechanism and prognostic value of activated AKT in pancreas cancer. Br J Cancer. 2003;89:2110-15
23. Guttilla IK, White BA. Coordinate regulation of FOXO1 by miR-27a, miR-96, and miR-182 in breast cancer cells. J Biol Chem. 2009;284:23204-16
24. Paik JH, Kollipara R, Chu G, Ji H, Xiao Y, Ding Z, Miao L, Tothova Z, Horner JW, Carrasco DR, Jiang S, Gilliland DG, Chin L, Wong WH, Castrillon DH, DePinho RA. FoxOs are lineage-restricted redundant tumor suppressors and regulate endothelial cell homeostasis. Cell. 2007;128:309-23
25. Narayan G, Pulido HA, Koul S, Vargas H, Zhang FF, Villella J, Schneider A, Terry MB, Mansukhani M, Murty VV. Frequent Promoter Methylation of CDH1, DAPK, RARB, and HIC1 Genes in Carcinoma of Cervix Uteri: Relationship to Clinical Outcome. Mol Cancer. 2003;2:24
26. Narayan G, Pulido HA, Nandula SV, Basso K, Sugirtharaj DD, Vargas H, Mansukhani M, Villella J, Meyer L, Schneider A, Gissmann L, Durst M, Pothuri B, Murty VV. Promoter Hypermethylation of FANCF: Disruption of Fanconi Anemia-BRCA Pathway in Cervical Cancer. Cancer Res. 2004;64:2994-97
27. Harris CP, Lu XY, Narayan G, Singh B, Murty VV, Rao PH. Comprehensive Molecular Cytogenetic Characterization of Cervical Carcinoma Cell Lines. Genes Chromosomes Cancer. 2003;36:233-41
28. Narayan G, Goparaju C, Pulido HA, Kaufmann AM, Schneider A, Durst M, Mansukhani M, Pothuri B, Murty VV. Promoter hypermethylation-mediated inactivation of multiple Slit-Robo genes in cervical cancer progression. Mol Cancer. 2006;5:16
29. Gardiner-Garden M, Frommer M. CpG islands in vertebrate genomes. J Mol Biol. 1987;196:261-82
30. Shankar S, Chen Q, Srivastava RK. Inhibition of PI3K/AKT and MEK/ERK pathways act synergistically to enhance antiangiogenic effects of EGCG through activation of FOXO transcription factor. J Mol Signal. 2008;3:7
31. Chen Q, Ganapathy S, Singh KP, Shankar S, Srivastava RK. Resveratrol Induces Growth Arrest and Apoptosis through Activation of FOXO Transcription Factors in Prostate Cancer Cells. PLoS ONE. 2010;5:e15288
32. Ma YY, Wei SJ, Lin YC, Lung JC, Chang TC, Whang-Peng J, Liu JM, Yang DM, Yang WK, Shen CY. PIK3CA as an oncogene in cervical cancer. Oncogene. 2000;19:2739-44
33. Narayan G, Bourdon V, Chaganti S, Pulido HA, Nandula SV, Rao PH, Gissmann L, Durst M, Schneider A, Pothuri B, Mansukhani M, Basso K, Chaganti RS, Murty VV. Gene Dosage Alterations Revealed by cDNA Microarray Analysis in Cervical Cancer: Identification of Candidate Amplified and Overexpressed Genes. Genes Chromosomes Cancer. 2007;46:373-84
34. Maekawa T, Maniwa Y, Doi T, Nishio W, Yoshimura M, Ohbayashi C, Hayashi Y, Okita Y. Expression and localization of FOXO1 in non-small cell lung cancer. Oncol Rep. 2009;22:57-64
35. Risinger JI, Maxwell GL, Chandramouli GV, Jazaeri A, Aprelikoya O, Patterson T, Berchuck A, Barrett JC. Microarray analysis reveals distinct gene expression profiles among different histologic types of endometrial cancer. Cancer Res. 2003;63:6-11
36. Myatt SS, Wang J, Monteiro LJ, Christian M, Ho KK, Fusi L, Dina RE, Brosens JJ, Ghaem-Maghami S, Lam EW. Definition of microRNAs that repress expression of the tumor suppressor gene FOXO1 in endometrial cancer. Cancer Res. 2010;70:367-77
37. Ward EC, Hoekstra AV, Blok LJ, Hanifi-Moghaddam P, Lurain JR, Singh DK, Buttin BM, Schink JC, Kim JJ. The regulation and function of the forkhead transcription factor, Forkhead box O1, is dependent on the progesterone receptor in endometrial carcinoma. Endocrinology. 2008;149:1942-50
38. Gilley J, Coffer PJ, Ham J. FOXO transcription factors directly activate bim gene expression and promote apoptosis in sympathetic neurons. J Cell Biol. 2003;162:613-22
39. Brunet A, Park J, Tran H, Hu LS, Hemmings BA, Greenberg ME. Protein kinase SGK mediates survival signals by phosphorylating the forkhead transcription factor FKHRL1 (FOXO3a). Mol Cell Biol. 2001;21:952-65
40. Qian J, Zou Y, Rahman J SM, Lu B, Massion PP. Synergy between phosphatidylinositol 3-kinase/Akt pathway and Bcl-xL in the control of apoptosis in adenocarcinoma cells of the lung. Mol Cancer Ther. 2009;8:101-9
Author contact
![]() Corresponding author: Gopeshwar Narayan, Cancer Genetics Laboratory, Department of Molecular and Human Genetics, Banaras Hindu University, Varanasi -221005, India. Phone no.: +91-542-670-2497 (office), +91-945-001-3701 (mobile); Fax: +91-542-670-2499 E-mail: gnarayanac.in.
Corresponding author: Gopeshwar Narayan, Cancer Genetics Laboratory, Department of Molecular and Human Genetics, Banaras Hindu University, Varanasi -221005, India. Phone no.: +91-542-670-2497 (office), +91-945-001-3701 (mobile); Fax: +91-542-670-2499 E-mail: gnarayanac.in.