Journal of Cancer
ISSN: 1837-9664
3.2
Impact Factor
ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2014; 5(8):633-645. doi:10.7150/jca.8466 This issue Cite
Research Paper
Gene Expression Analysis Reveals Distinct Pathways of Resistance to Bevacizumab in Xenograft Models of Human ER-Positive Breast Cancer
Yesim Gökmen-Polar1 ![]() , Chirayu P. Goswami2*, Rachel A. Toroni3, Kerry L. Sanders3, Rutika Mehta1, Usha Sirimalle1, Bogdan Tanasa4, Changyu Shen2, Lang Li2, Mircea Ivan3, Sunil Badve1,3, George W. Sledge Jr1,3
, Chirayu P. Goswami2*, Rachel A. Toroni3, Kerry L. Sanders3, Rutika Mehta1, Usha Sirimalle1, Bogdan Tanasa4, Changyu Shen2, Lang Li2, Mircea Ivan3, Sunil Badve1,3, George W. Sledge Jr1,3 ![]() #
#
1. Department of Pathology and Laboratory Medicine, Indiana University School of Medicine, Indianapolis, IN;
2. Center for Computational Biology and Bioinformatics, Indiana University School of Medicine, Indianapolis, IN;
3. Department of Medicine, Indiana University School of Medicine, Indianapolis, IN;
4. Scripps Research Institute, University of Medicine and Pharmac, La Jolla, CA.
# Current address: Department of Medicine, Stanford University, Palo Alto, CA.
* Current address: Thomas Jefferson University Hospitals, Philadelphia, PA.
Received 2014-1-1; Accepted 2014-6-20; Published 2014-8-15
Citation:
Gökmen-Polar Y, Goswami CP, Toroni RA, Sanders KL, Mehta R, Sirimalle U, Tanasa B, Shen C, Li L, Ivan M, Badve S, Sledge Jr GW. Gene Expression Analysis Reveals Distinct Pathways of Resistance to Bevacizumab in Xenograft Models of Human ER-Positive Breast Cancer. J Cancer 2014; 5(8):633-645. doi:10.7150/jca.8466. https://www.jcancer.org/v05p0633.htm
Other stylesAbstract
Bevacizumab, the recombinant antibody targeting vascular endothelial growth factor (VEGF), improves progression-free but not overall survival in metastatic breast cancer. To seek further insights in resistance mechanisms to bevacizumab at the molecular level, we developed VEGF and non-VEGF-driven ER-positive MCF7-derived xenograft models allowing comparison of tumor response at different timepoints. VEGF gene (MV165) overexpressing xenografts were initially sensitive to bevacizumab, but eventually acquired resistance. In contrast, parental MCF7 cells derived tumors were de novo insensitive to bevacizumab. Microarray analysis with qRT-PCR validation revealed that Follistatin (FST) and NOTCH were the top signaling pathways associated with resistance in VEGF-driven tumors (P<0.05). Based on the presence of VEGF, treatment with bevacizumab resulted in altered patterns of metagenes and PAM50 gene expression. In VEGF-driven model after short and long-term bevacizumab treatments, a change in the intrinsic subtype (luminal to myoepithelial/basal-like) was observed in association with increased expression of genes implicated with cancer stem cell phenotype (P<0.05). Our results show that the presence or absence of VEGF expression affects the response to bevacizumab therapy and gene pathways. In particular, long-term bevacizumab treatment shifts the cancer cells to a more aggressive myoepithelial/basal subtype in VEGF-expressing model, but not in non-VEGF model. These findings could shed light on variable results to anti-VEGF therapy in patients and emphasize the importance of patient stratification based on the VEGF expression. Our data strongly suggest consideration of patient subgroups for treatment and designing novel combinatory therapies in the clinical setting.
Keywords: Bevacizumab, vascular endothelial growth factor, breast cancer, estrogen receptor, de novo and acquired resistance.
Introduction
Vascular endothelial growth factor (VEGF) is a well-known stimulator of tumor angiogenesis, growth and metastasis in breast cancer. Overexpression of VEGF is an early event in breast cancer progression and a prerequisite step to tumor invasion (e.g., in ductal carcinomas in situ) [1]. VEGF and its receptors have been identified in different breast cancer subtypes [2-4]. Elevated expression of VEGF can be associated with shorter relapse-free survival and overall survival times in breast cancer patients with both positive and negative lymph nodes [3, 4]. Recently, higher intratumor levels of VEGF have also been reported in triple receptor-negative breast cancer and are associated with a shorter overall survival duration [5]. Overexpression of VEGF has also been linked to increased metastatic potential, resistance to chemotherapy and hormonal therapy [6-8]. Therefore, therapeutic targeting of VEGF should have promise in breast cancer.
The humanized monoclonal antibody against VEGF, bevacizumab ([BEV] Avastin; Genentech Inc., South San Francisco, CA), is the most advanced anti-VEGF therapy in breast cancer. While the first phase III trial of BEV in refractory metastatic breast did not provide a benefit in combination with capecitabine [9], the E2100 trial demonstrated significant prolongation of the median progression-free survival in metastatic breast cancer [10]. Additionally, this trial identified an association of the VEGFA-2578 AA and -1154 AA genotypes with better median overall survival than other genotypes [11]. Two subsequent phase III trials (AVADO and Ribbon-1) in the first-line setting also demonstrated improvements in progression-free survival [12, 13]. Although these trials met the primary endpoint of progression-free survival, they did not show an improvement in overall survival.
One obvious limitation of these trials has been the lack of therapeutic individualization; i.e., stratification of patients based on VEGF expression. Another note of caution issued by preclinical studies suggested that adjuvant anti-VEGF therapies may increase the risk of metastasis [14, 15], although this fact has not been observed in clinical settings. The mixture of promises and partial clinical failures prompts further efforts to gain a deeper understanding of the tumor response and adaptation to the anti-VEGF challenge and develop rational strategies to overcome resistance to such agents.
In this study, we aimed to investigate the mechanisms of resistance to BEV in breast cancer. To this end, we developed in vivo breast cancer models of both short term and long term responses to BEV, based on a non-VEGF and VEGF-expressing MCF7 cells.
Here, we report distinct pathways of response to BEV in these models, which may provide critical insights into the alteration of breast cancer biology during VEGF blockade. These pathways may also provide important clues for identifying synergistic therapeutic strategies to overcome or delay resistance to antiangiogenic agents.
Materials and Methods
Breast cancer cell lines
ML20 (MCF7 cells transfected with control plasmid and MV165 (MCF7 cells transfected with VEGF) were a kind gift from Dr. F. G. Kern [16]. Both cell lines have been carefully maintained in a humidified tissue culture incubator at 37 oC in 5% CO2, and stocks of the earliest passage cells have been stored. The cell lines were grown in culture media containing supplements as described previously in the reference.
Orthotopic xenograft models
MV165 tumors display a better tumor engraftment compared with ML20. Tumor characteristics have been characterized previously [17]. ML20 (10×106) or MV165 cells (5×106) were implanted into the mammary fat pad of athymic nude mice as described previously [17, 18]. Once tumors were established, the mice were treated with the saline control (PBS) or BEV (5mg/kg, i.p./twice weekly). Treatment was continued for 3 weeks when the tumors are sensitive to BEV at an early timepoint (defined as the group of sensitive to BEV, short-term [ST]). We continued to treat mice with vehicle, BEV, or anti-mouse VEGF antibody to develop resistant tumors, and harvested tumors when they reached the tumor burden limit (2000mm3). At 8 weeks, they were harvested when they were progressing and insensitive to BEV (defined as the group of resistant, long-term [LT]). In addition, mice were treated with anti-mouse VEGF antibody (Genentech, 5mg/kg, i.p./twice weekly) for both early and late timepoints (n=6 per each condition). All tumors were harvested at the designated timepoints and snap frozen in liquid nitrogen and stored at -80 oC until RNA isolation. Tumor volume was calculated as L×W2/2, where L is length and W is width. All animal experiments were done under a protocol approved by the Indiana University Institutional Animal Care and Use Committee.
Microarray analysis
Total RNA was extracted from snap-frozen tumors using TRIzol Reagent method (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions. The quality of RNA was assessed using the Nanodrop ND-1000 Spectrophotometer (ThermoScientific, Wilmington, DE). The RNA integrity number was measured using the Agilent Bioanalyzer (Agilent Technologies, Santa Clara, CA). All RNA samples were treated with Turbo DNase (Ambion, Foster City, CA). 200ng of total RNAs from tumors sensitive to BEV (3 weeks; ST) or resistant to BEV (8 weeks; LT) or with vehicle control group were subjected to whole-genome gene expression analysis for both human WG-6v2 and mouse WG-6v2 Expression Beadchips (Illumina, San Diego, CA) as per manufacturer's protocols (n=4 per each condition).
Data preprocessing
Data was analyzed for each condition in triplicates from Illumina human and mouse WGGEX arrays. Genes which had a poor signal quality across a maximal number of arrays were filtered out. As a result, 10,804 genes and 15,492 genes were found to have signals significantly above background for human and mouse arrays, respectively. The data was quantile normalized and log2 transformed before statistical analysis. We performed one-way ANOVA analysis to identify differentially expressed genes in our dataset.
Ingenuity Pathways Analysis
To identify the statistically significant biological functions, and signaling pathways affected by the genes differentially expressed in our comparisons, we performed Ingenuity Pathways Analysis (IPA; Ingenuity Systems, Inc). IPA is the largest curated database and analysis system for understanding the signaling and metabolic pathways, molecular networks, and biological processes that are most significantly changed in a dataset of interest (http://www.ingenuity.com).
Validation of the selected genes by real-time quantitative reverse transcription PCR (qRT-PCR)
All qRT-PCR reactions were performed in triplicates. Total RNAs were reverse-transcribed using high capacity cDNA reverse transcription kit. The mRNA levels were analyzed by real-time qRT-PCR using TaqMan gene expression assays on an ABI Prism 7900 platform according to the manufacturer's instructions (Applied Biosystems, Foster City, CA). Actin (ACTB) was used as endogenous control for normalization purpose. The relative quantification of the gene expression changes (fold) was analyzed according to DDCt method using the Applied Biosystems DataAssistTM Software v3.0.
Immunohistochemical staining
Tumors per condition (triplicates) were fixed with formalin for immunohistochemistry. Immunostaining procedures for keratin 5/6 (KRT5/6 or cytokeratin 5/6), keratin 14 (KRT14 or cytokeratin 14), ER, and Ki67 were performed as described previously [19-21]. Images were recorded using an Olympus BS41 microscope with DP72 camera.
Results
Xenografts exhibit different characteristics of resistance to BEV treatment based on the presence of VEGF expression status
To investigate the mechanisms associated with resistance to BEV therapy, we first compared tumor response at different timepoints of BEV treatment. Tumor growth was supported with estrogen, as described before [16]. Non-VEGF xenograft tumors (ML20) exhibited no visible decrease in growth during VEGF blockade, suggesting de novo resistance (Fig. 1A). In contrast, BEV significantly reduced the primary tumor growth of the VEGF-overexpressing xenograft tumors (MV165) at 3 weeks (5mg/kg, i.p./twice weekly) (Fig. 1B). Despite the initial response to BEV, MV165 tumors showed development of acquired resistance and progressed to the tumor volume of control mice after prolonged (8 weeks) treatment. Additional mice were treated with murine anti-VEGF monoclonal antibody (muMAb VEGF; Genentech; 5mg/kg, i.p./twice weekly) to confirm that the resistance was not due to murine VEGF of stromal origin. muMAb VEGF inhibited the tumor growth in MV165 xenograft tumors in a similar fashion (data not shown).
Gene expression profiles - human-specific arrays
VEGF-driven MV165 and non-VEGF-driven ML20 xenograft models
To identify the molecular mechanisms associated with the acquired resistance to BEV therapy, we performed gene expression analysis for VEGF-driven MV165 tumors treated with BEV at 3 weeks sensitive to therapy (ST treatment; MV165-ST) and 8 weeks resistant to therapy (LT treatment; MV165-LT) using Illumina human-specific (WG-6 v2) whole-genome expression arrays. Control tumors treated with vehicle were also analyzed (MV165-V [vehicle]). Differentially regulated genes were determined using ANOVA and IPA [13]. Of the 10,894 genes that were significantly above background, 64 and 37 genes were differentially regulated between MV165-LT versus MV165-ST and MV165-LT versus MV165-V, respectively (ANOVA) (Additional File 1: Suppl. Fig. S1). Among the 64 genes in MV165-LT compared with MV165-ST, 61 genes were unique to tumor growth after initial BEV response, suggesting that these genes mediate acquired resistance. Twenty-four genes were altered when BEV reduced the tumor growth significantly at 3 weeks. Among the 24 genes, 12 genes were unique to MV165-ST sensitive group. Using IPA, Table 1 illustrates the top five differentially regulated genes in MV165-LT and MV165-ST groups (P ≤0.05). Follistatin (FST), the activin/bone morphogenetic protein (BMP) antagonist, and HEY2, the downstream effector of NOTCH pathways, were among the top genes upregulated in acquired resistance to anti-BEV therapy. These results indicate that LT BEV treatment alters alternative biological pathways that may be associated with acquired tumor resistance.
Due to the lack of therapeutic individualization in the clinic, we also aimed to investigate the mechanisms of resistance to BEV in ML20 xenograft tumors, a non-VEGF and ER-positive breast cancer preclinical model. As expected, ML20 tumors lacking VEGF were insensitive to BEV even at ST treatment representing de novo resistance (relative primary resistance). Of the 10,894 genes found to have signals significantly above background, 40 and 67 genes were differentially regulated in ML20-ST versus ML20-V and ML20-ST versus ML20-LT, respectively (Additional File 1: Suppl. Fig. S2). Of the 40 genes in ML20-ST compared with ML20-V, 30 genes were altered unique to ST BEV treatment, while 23 of 38 genes in ML20-LT compared with ML20-V were altered after LT treatment to BEV. Comparison of LT versus ST further revealed that 49 genes were altered in LT rather than ST. Using IPA, Table 1 illustrates the top five differentially regulated genes in ML20-ST and ML20-LT groups (P≤0.05). MUC5AC/MUC5B and HEY2 were among the top genes, suggesting a role for IL17 signaling, and NOTCH pathways in de novo resistance to BEV therapy. In addition, other top pathways altered include G-protein coupled receptor signaling. Interferon signaling is activated in ST and LT treatment when compared with vehicle.
Figure 1
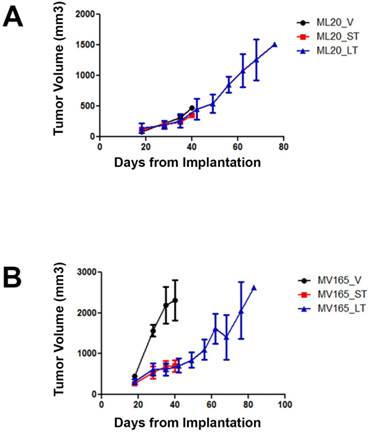
ML20 (non-VEGF-driven MCF7) and MV165 (VEGF-driven MCF7) xenografts exhibit different patterns of response to BEV treatment at different timepoints. MV165 (5x106) and ML20 (10x106) cells were implanted into mammary fat pads of athymic mice (4 mice per group) in the presence of supplemental estrogen. Treatment with BEV (5mg/kg/ i.p/twice weekly) or vehicle control was started when tumors were palpable. A, ML20-V, ML20-ST, ML20-LT, (n=4), B, MV165-V, MV165-ST, MV165-LT. BEV, bevacizumab; LT, long-term; ST, short-term; V, vehicle.
Table 1
Top differentially-regulated genes in MV165 and ML20 in response to short-term (ST) and long-term (LT) bevacizumab treatment using human arrays.
| Gene Symbol | Canonical Pathway | Gene Symbol | Canonical Pathway |
|---|---|---|---|
| MV165-LT versus MV165-ST | ML20-LT versus ML20-ST | ||
| FST | TGFB/BMP signaling, regulator of angiogenesis | IF44 | Interferon signaling |
| HEY2 | NOTCH signaling | IF127 | Interferon signaling |
| FGD3 | CDC42 signaling | IFI544L | Interferon signaling |
| MUCL1 | Bone micrometastasis | PARP10 | Retinoic acid mediated apoptosis signaling |
| CYP26B1 | RAR activation | BGN | BMP signaling |
| MV165-LT versus MV165-V | ML20-LT versus ML20-V | ||
| HBA1/HBA2 | Oxygen transport | IFI44 | Interferon signaling |
| CYP26B1 | RAR activation | MUC5AC | IL17A signaling in airway cells |
| PCP4 | B cell tolerance | IFI27 | Interferon signaling |
| AOX1 | NRF2-mediated oxidative stress response, amino acid metabolisms | PARP10 | Retinoic acid mediated apoptosis signaling |
| APOD | RAR activation | HEY2 | NOTCH signaling |
| MV165-ST versus MV165-V | ML20-ST versus ML20-V | ||
| HBA1/HBA2 | Oxygen transport | MUC5AC/MUC5B | IL17A signaling in airway cells |
| AGR3 | Steroid hormones regulation | HEY2 | NOTCH signaling |
| APOD | RAR activation | GALR2 | G-protein coupled receptor signaling |
| AOX1 | NRF2-mediated oxidative stress response, amino acid metabolisms | AGTR1 | G-protein coupled receptor signaling |
| AGR2 | Steroid hormones regulation | ARL4A | IL17A signaling in airway cells |
* ANOVA and Ingenuity Pathway Analysis have been performed; P≤0.05
†Fold change ≥1.5 or ≤-1.5
Real-time qRT-PCR validation of the differential expression for the selected genes in response to ST and LT treatment to BEV
The top differentially expressed genes in VEGF-driven model associated with acquired resistance to BEV were validated by qRT-PCR. The expression of FST was decreased 3-fold in the sensitive group and was increased 2.90-fold in the resistant VEGF-driven MV165 group compared with vehicle confirming the expression levels observed in microarray analysis (Fig. 2). BEV altered the mRNA expression levels of FST in an inverse manner; namely upregulating it in resistance group and downregulating it in sensitive group. We next assessed two members of NOTCH signaling, NOTCH3 and NOTCH4 receptors, whose altered expression levels have been implicated in the commitment of bipotent progenitors to the luminal lineage in normal human mammary development [22]. NOTCH4 levels were elevated in both sensitive and resistant groups to BEV (2.48-fold and 5-fold, respectively), but was statistically significant only in the resistant group (P<0.05). NOTCH3 expression was similar in these groups. In contrast to FST expression, levels of NOTCH4 were gradually increased, resulting in a significant upregulation in resistant group. In contrast, BEV treatment did not alter the gene levels in the same manner in ML20 (non-VEGF) model. There was a trend that the levels of FST and NOTCH4 were elevated in the LT, but these results did not reach the statistical significance as in MV165 (VEGF-driven) model, supporting the differential regulation of top pathways (FST, NOTCH3, and NOTCH4) between two models.
Figure 2
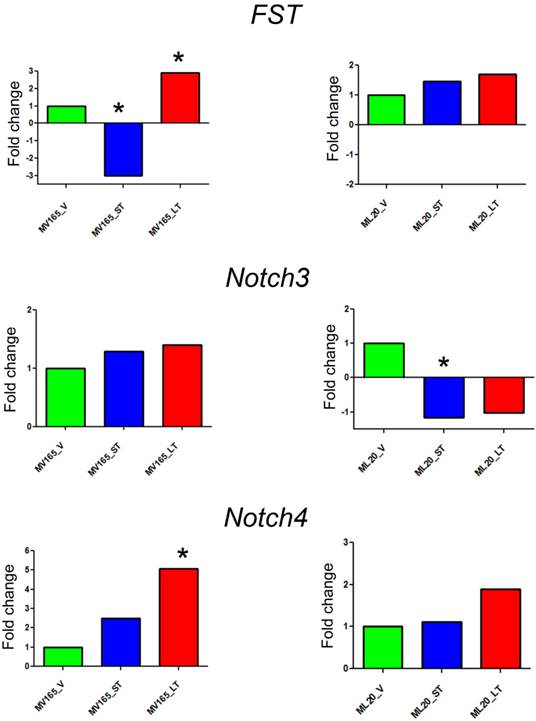
Validation of selected genes using quantitative real-time RT-PCR. Fold change for each comparison was calculated using DataAssist Software (Applied Biosystems, Foster City, CA). Data is normalized to a reference gene (ACTB). P<0.05 is considered significant. ML20 (non-VEGF) and MV165 (VEGF-expressing); *P<0.05 statistically significant. LT, long-term; ST, short-term; V, vehicle.
Comparison of gene expression profiles of ST and LT treatment of BEV with the eight metagenes in VEGF-drive and non-VEGF-driven breast cancer models
To assess the impact of short term and sustained BEV treatment on clinically relevant pathways, we investigated gene expression in eight metagenes of significant relevance for breast cancer [23]. Comparison of the selected metagenes demonstrated that BEV profoundly decreased the genes in the proliferation metagene in MV165 xenografts (Fig. 3A). However, most of the basal cytokeratins in basal-like metagene were upregulated in response to BEV regardless of treatment duration. Comparisons of ML20 (non-VEGF model) gene profiling with metagenes revealed distinct differences between ML20 and MV165 in response to BEV treatment (Figs. 3A, 3B). Unlike the MV165 model, BEV upregulated some of the proliferation genes in ML20 model (Fig. 3B).
Figure 3
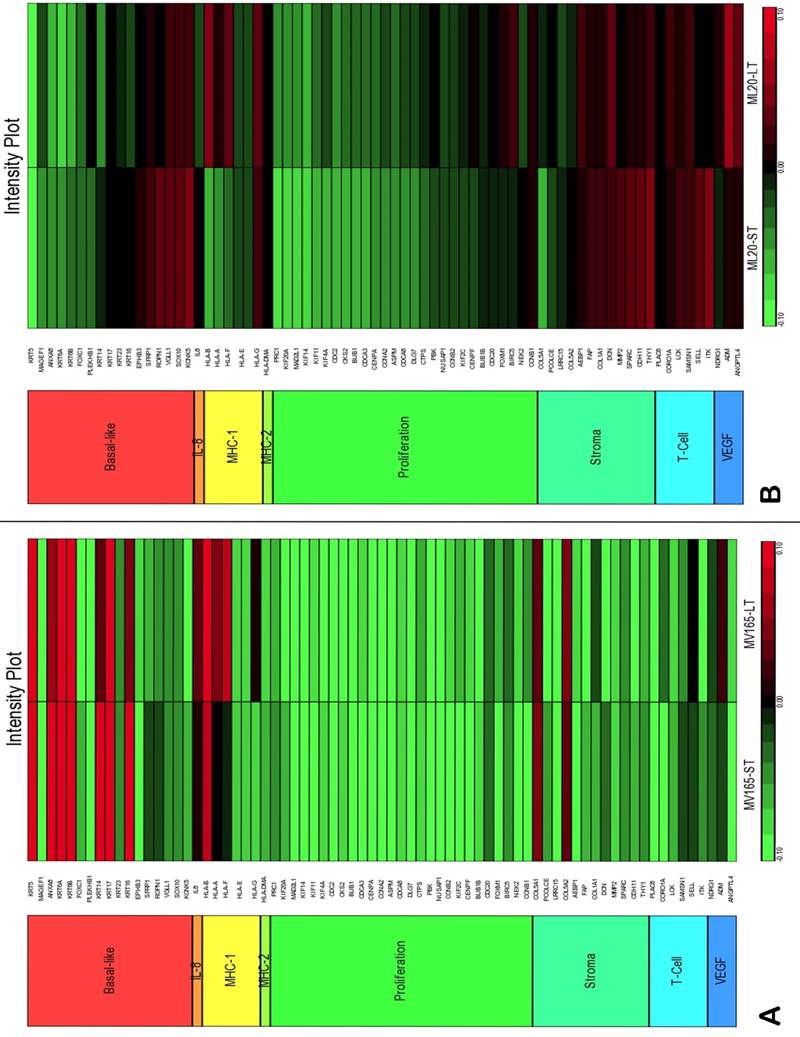
Comparison of gene expression profiles of short-term (ST) and long-term (LT) treatment of bevacizumab with the eight metagenes in VEGF-driven and non-VEGF-driven breast cancer models. Heatmap representation of differentially regulated genes in comparison to eight metagenes A, MV165-ST and MV165-LT normalized to vehicle, B, ML20-ST and ML20-LT normalized to vehicle, C, comparison of MV165-ST and MV165-LT normalized to vehicle to hypoxia metagene, D, comparison of ML20-ST and ML20-LT normalized to vehicle to hypoxia metagene.
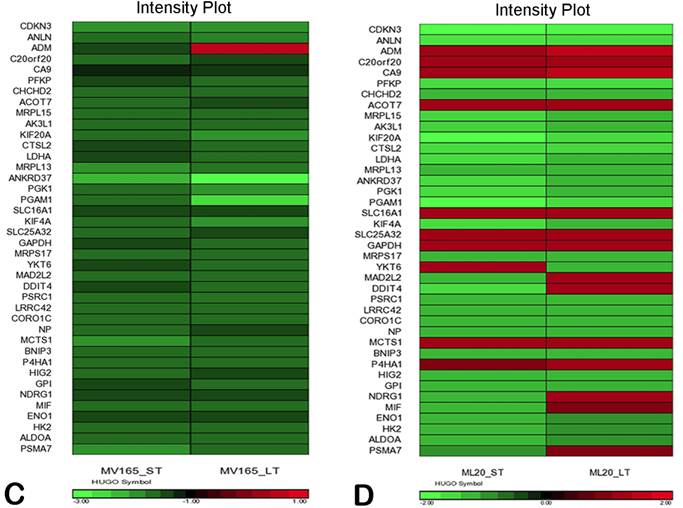
We also compared the effect of BEV on other metagenes representative of adaptive responses to the tumor microenvironment. Of the 12 differentially regulated genes in the “stromal metagene,” BEV downregulated all genes except COL5A1 and COL5A2 (Fig. 3A). Of the six differentially regulated genes in T-cell metagene, BEV reduced the expression levels of all five genes except SELL, while genes in MHC1 metagene were upregulated particularly in response to LT treatment. Among the VEGF metagene, the expression levels of NDRG1 and ANGPTL4 were reduced in both ST and LT, but levels of ADM were increased in response to LT treatment. Most of the genes in VEGF metagene were regulated by HIF1A gene. However, we did not observe upregulation of hypoxia markers except ADM. Analyzing the hypoxia metagene [24], we further confirmed that the genes in hypoxia metagene exhibited low expression in response to both ST and LT of BEV except ADM in VEGF-driven model (Fig. 3C). Among the other metagenes, it was notable that IL8 was gradually increased in response to BEV treatment, more significantly in the LT treatment (Fig. 3A).
In contrast to MV165 xenograft tumors, genes within the stromal metagene, T-cell and VEGF metagenes in ML20 were significantly upregulated in response to BEV treatment at both short and long treatment to BEV (Fig. 3B). On the other hand, genes in MHC1 metagene were downregulated, which was the opposite response when compared with MV165. Another distinct observation was that some of the hypoxia genes, including CA9, SLC16A1, NDGR1, and GAPDH, were upregulated in ML20, suggesting that BEV induces hypoxia markers in the non-VEGF model in contrast to MV165 (Fig. 3D).
BEV treatment and altered intrinsic subtypes in VEGF-driven tumors
Using DNA microarrays, Perou et al [25, 26] and Sorlie et al [27] classified breast carcinomas into five molecular subtypes: ER-positive (luminal A), ER-positive (luminal B), ER-negative/HER2-enriched, basal-type, and normal-like subtypes. A 50-gene subtype predictor assay (PAM50) has been developed representing each intrinsic subtype of breast cancer [28]. We analyzed the expression levels of the PAM50 genes in microarray analysis of MV165 samples. Thirty-six of these 50 genes showed differential regulation in response to ST and/or LT treatment to BEV compared with vehicle (Fig. 4A).
Figure 4
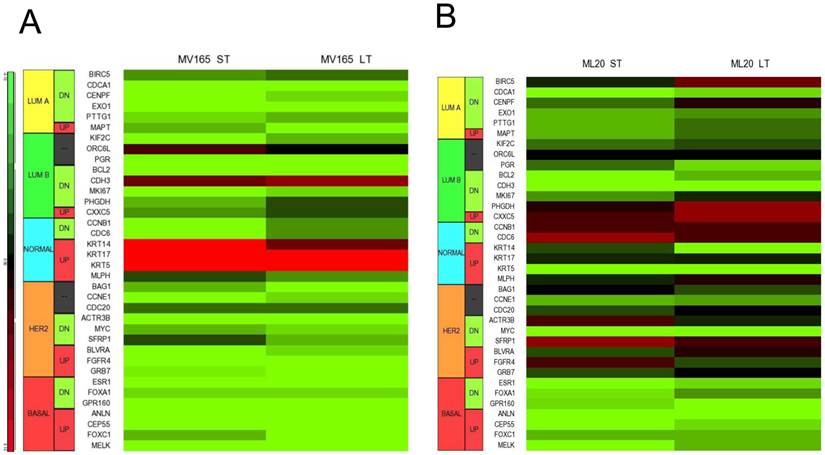
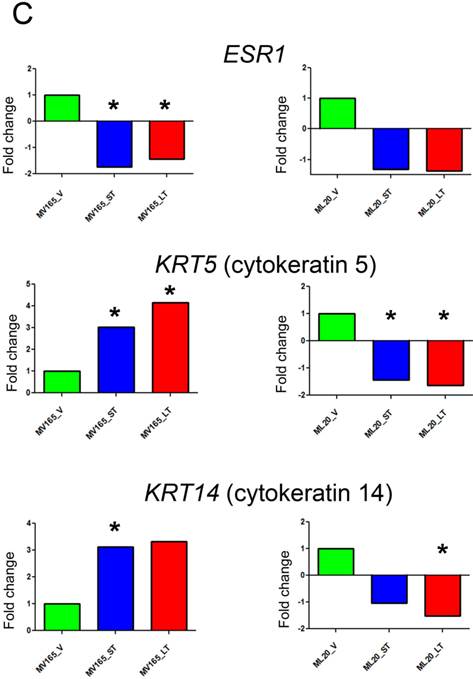
Effect of bevacizumab on intrinsic subtypes in ML20 and MV165 xenograft tumors; Heatmap representation of differentially regulated genes in comparison to PAM50 genes A, MV165-ST (short-term) and MV165-LT (long term) normalized to vehicle, B, ML20-ST and ML20-LT normalized to vehicle, C, Quantitative real-time RT-PCR validation of ESR1, KRT5 (cytokeratin 5), and KRT4 (cytokeratin 14). Fold change for each comparison was calculated using DataAssist Software (Applied Biosystems). Data is normalized of a reference gene (ACTB). *P<0.05 statistically significant.
As expected, most of the genes representative of low proliferation in luminal A (BIRC5, CENPF, PTTG1) were downregulated in response to BEV irrespective of the treatment period. Furthermore, a shift to luminal B from luminal A after development of resistance to BEV treatment was not observed. Markers in luminal B, such as BCL2 and MKI67, were decreased in both ST and LT treatment. However, few genes were elevated, such as CDH3, KRT14 (CK14), and KRT5 (CK5), in response to BEV. The upregulation of basal-like cytokeratins (CK5/6 and CK14) was confirmed by comparison of PAM50 genes. In contrast, ESR1 (estrogen receptor alpha) is downregulated in both ST and LT BEV treatments.
We next examined the expression levels of PAM50 genes in ML20 microarray sets. As in MV165 model, 36 of these 50 genes showed differential regulation in response to ST and/or LT treatment to BEV compared with vehicle (Fig. 4B). However, a distinct set of genes were differentially regulated in ML20 compared with MV165 groups. The genes upregulated showed a more complex gene expression profiling including BIRC5, PHGDH, CDC6, SFRP1, and FGFR4. ESR1, KRT5, and KRT14 were downregulated in this set. In general, BEV treatment showed a more complex gene expression profiling in comparison to intrinsic subtypes.
To further validate the effect of BEV on intrinsic subtypes, we performed qRT-PCR using the representative genes for luminal A and myoepithelial gene sets (ESR1, KRT5, and KRT14) (Fig. 4C). ESR1 showed a significant decrease in both sensitive and resistant group compared with the vehicle of VEGF-driven tumor (P<0.05). On the other hand, KRT5 (CK5) levels were increased significantly in both sensitive (3.02-fold; P<0.05) and resistant (4.14-fold; P<0.05) groups, while KRT14 showed a 3.12-fold increase (P<0.05) in sensitive and a 3.33-fold increase in resistant group compared with the vehicle group. These results suggest that BEV treatment may contribute to a switch of tumor cells with luminal characteristics to a myoepithelial, basal-like phenotype independent of treatment duration.
In non-VEGF ML20, downregulation of ESR1 was not significant. In addition, KRT5 and KRT14 were downregulated in both ST and LT treatments. KRT5 levels were significantly reduced in ST (-1.66-fold; P<0.05) and LT (-2.12-fold; P<0.05), while KRT14 downregulation was significant at the LT (-1.96-fold; P<0.05).
Effect of BEV on intrinsic subtypes at the protein level
To further validate the altered representative genes of intrinsic subtypes (ESR1, KRT5, and KRT14) at the protein level, we next performed immunostaining for these genes. We confirmed the upregulation of KRT14 in BEV-resistant MV165 cells (P=0.02), whereas the ML20 (de novo resistance) model presented a significant decrease in KRT14 which confirmed the observations at the mRNA level (P=0.047) (Fig. 5A). We did not observe expression changes at the significant level for other genes (KRT5 and ESR1) at the protein level (data not shown).
Figure 5
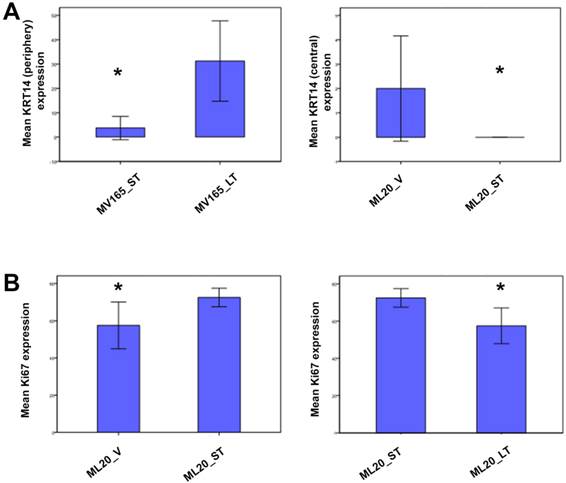
Validation of selected genes using immunohistochemistry. ML20 (non-VEGF) and MV165 (VEGF-expressing); *Mann-Whitney test P<0.05 statistically significant. LT, long-term; ST, short-term; V, vehicle.
To determine the proliferative effect of BEV on ML20 as observed by the PAM50 analysis, we assessed the immunostaining for Ki67, the most widely used proliferation marker in breast tumor samples [29]. As expected, Ki67 expression was higher in ML20-ST tumors compared with ML20-V being statistically significant (P=0.044) (Fig. 5B). The Ki67 expression was lowered with the longer treatment of BEV [ML20-LT; P=0.044)]. These results suggest the distinct effect of BEV in the presence or absence of VEGF overexpression.
Gene expression analysis of tumor environment using mouse-specific arrays
To determine additional mechanisms associated with tumor microenvironment (e.g., stroma, endothelial cells, and pericytes of mouse origin) in ML20 and MV165 xenografts, we performed mouse specific gene expression profiling (mouse WG-6 v2.0 Expression Beadchips, Illumina) using V-, ST-, and LT-treated tumors for MV165 and ML20 xenografts. We observed that a larger number of stromal mouse genes were significantly altered in BEV progression versus progression with human tumor genes in MV165 tumors (Additional File 1: Suppl. Figs. S3, S4). Of the 15,492 genes that were expressed above background, 58 and 417 genes were differentially regulated between MV165-LT versus MV165-ST and MV165-LT versus MV165-V, respectively (P≤0.001) (Additional File 1: Suppl. Fig. S3). Among the 58 genes in MV165-LT compared with MV165-ST, 35 genes were unique to LT BEV, suggesting candidate genes for acquired resistance. In contrast, 294 genes were altered when BEV reduced the tumor growth significantly at 3 weeks. Among these 294 genes, 81 genes were unique to MV165-ST sensitive group. Table 2 illustrates the top differentially regulated genes in MV165-LT and MV165-ST groups using mouse-specific arrays (P≤0.001). The top significant upregulated genes in resistant MV165-LT compared with sensitive MV165-ST were SLPI, GREM1, CCL21, CCL5 (RANTES), and CXCR6. Among the canonical pathways associated with these genes, most differentially regulated pathways were immune system-related signaling such as altered T cell and B cell signaling in rheumatoid arthritis, communication between innate and adaptive immune cells, and CCR5 signaling in macrophages. Other pathways included glucocorticoid receptor signaling, TGFB/BMP/hedgehog signaling, G-protein coupled receptor signaling, and AKT/mTOR/S6K signaling. Taken together, these results suggest that LT BEV treatment may alter the immune and the inflammatory response that needs to be verified with other systems. ST treatment (MV165-ST versus MV165-V) suggests differential regulation of TGFB pathway members such as TGFB1.
We next compared ML20 tumors insensitive at the shorter treatment time with vehicle-treated tumors using mouse-specific arrays. Of the 15,492 genes that were above background, 542 and 22 genes were differentially regulated in ML20-ST compared with ML20-V and ML20-ST compared with ML20-LT, respectively (P≤0.001) (Additional File 1: Suppl. Fig. S4). Table 2 lists the top differentially regulated genes in ML20-ST and ML20-LT groups using mouse arrays (P≤0.001). The top significant downregulated genes in ML20-ST compared with ML20-V were SPP1, F2R, COL4A2, LGMN, and CTGF. No upregulated genes were present at P≤0.001. The top canonical pathways associated with these genes include the role of osteoblasts, osteoclasts and chondrocytes, clathrin-mediated endocytosis signaling/glioma invasiveness signaling and T cell regulation. Further comparison of ML20 tumors with LT treatment versus ST treatment have identified the germ cell-Sertoli cell junction signaling, glucocorticoid receptor signaling and immune response pathways among the top pathways. These results suggest that the nature of response and resistance is primarily altered based on the presence or absence of VEGF expression.
Table 2
Top differentially-regulated genes in MV165 and ML20 in response to short-term (ST) and long-term (LT) bevacizumab treatment using mouse arrays
| Gene Symbol | Canonical Pathway | Gene Symbol | Canonical Pathway |
|---|---|---|---|
| MV165-LT versus MV165-ST | ML20-LT versus ML20-ST | ||
| SLPI | Tumor angiogenesis/ glucocorticoid receptor signaling | ACTG2 | Germ cell-Sertoli cell junction signaling |
| GREM1 | TGFB/BMP/hedgehog signaling | SLPI | Tumor angiogenesis/ glucocorticoid receptor signaling |
| CCL21 | Altered T cell and B cell signaling in rheumatoid arthritis | EGR1 | Immune response pathways |
| CCL5 (RANTES) | Communication between innate and adaptive immune cells/ CCR5 signaling in macrophages/ glucocorticoid receptor signaling/ chemokine signaling | BCKDK | Glucocorticoid receptor signaling |
| CXCR6 | G-protein coupled receptor signaling/ AKT/mTOR/S6K signaling | ANKRD1 | 26S proteasome pathway |
| MV165-LT versus MV165-V | ML20-LT versus ML20-V | ||
| CXCL14 | Dendritic cell maturation/glucose metabolism | MGP | BMP pathway |
| GREM1 | TGFB/BMP/hedgehog signaling | SPP1 | Role of osteoblasts, osteoclasts and chondrocytes |
| CD72 | Systemic lupus erythematosus signaling | B2M | Regulation of immune response |
| CD52 | T cell activation | LOX | Lipoxygenase and cyclooxygenase pathways |
| LY6E | Interferon signaling; systemic lupus erythematosus signaling | F2R | Clathrin-mediated endocytosis signaling/glioma invasiveness signaling |
| MV165-ST versus MV165-V | ML20-ST versus ML20-V | ||
| CD72 | Systemic lupus erythematosus signaling | SPP1 | Role of osteoblasts, osteoclasts and chondrocytes |
| CH25H | Cholesterol and lipid metabolism | F2R | Clathrin-mediated endocytosis Signaling/glioma invasiveness signaling |
| TGFBI | TGFB signaling | COL4A2 | |
| ESM1 | Chemokine signaling | LGMN | T cell regulation |
| COL4A1 | Intrinsic Prothrombin activation pathway | CTGF | Hepatic fibrosis/hepatic stellate cell activation |
* ANOVA and Ingenuity Pathway Analysis have been performed; P value ≤0.005
†Fold change ≥1.5 or ≤-1.5
Discussion
Despite the initial promises, patients with breast cancer treated with BEV therapy have only a transitory benefit. This might be attributed to the lack of identification of select groups of patients who might benefit from this drug. Furthermore, numerous potential mechanisms may contribute to resistance to antiangiogenic agents including VEGF therapy (reviewed in Sledge et al [30]). It will be important to understand and overcome the underlying mechanisms of resistance. Therefore, clinically relevant preclinical models are critical to examine the potential mechanisms of antiangiogenic resistance at the molecular level. Novel targets identified in these models are potential candidates that need to be validated in clinical studies.
MCF7 breast cancer xenografts overexpressing VEGF (MV165) have been shown to exhibit greater tumor growth and result in acquisition of metastatic capability into the lungs of the orthotopically implanted xenograft-bearing mice, while VEGF-lacking (ML20) breast cancer xenografts have a much slower growth rate and do not metastasize [16, 31]. ML20 xenograft tumors that lack expression of VEGF exhibited de novo resistance to BEV. In contrast, MV165 cells were sensitive to BEV treatment. However, this effect was transitory, as the tumors grow back even after continuation of the therapy, indicating development of acquired resistance. Given the fact that ML20 and MV165 differ by genetically overexpressed VEGF, this indicates that BEV may exert its anti-tumor activity mainly through its actions on VEGF ligand and receptor pathway and the presence or absence of VEGF determines the nature of resistance, de novo or acquired, to BEV.
Comparative analysis of human-specific and mouse-specific microarrays revealed that MV165 and ML20 xenograft tumors exhibited distinct gene profiles (Additional File 1: Suppl. Figs. S1-S4). Using human-specific arrays, the TGFB/BMP/hedgehog signaling was among the top significant genes in MV165 set. The expression levels of FST gene were inversely correlated with BEV sensitivity both in microarray and qRT-PCR analyses. FST, the antagonist of activin belonging to TGFB superfamily, acts as a pleiotropic growth factor system that controls cell proliferation, differentiation, and apoptosis in several models in an autocrine and paracrine fashion [32-34]. The role of FST is complex and tissue context dependent. Krneta et al. found that FST-expressing tumors were smaller in human R30C mammary carcinoma model compared with activin-expressing tumors [35]. In contrast, in our study, FST expression was associated with proliferation; inhibition of tumor growth was correlated with inhibition of FST levels.
Upregulation of genes involved in NOTCH and WNT signaling was also observed in BEV resistant MV165 tumors. HEY2, a downstream effector of NOTCH signaling, was among the top significantly upregulated genes. Transcriptome profiling of normal human mammary subpopulation has revealed the expression of FST and NOTCH4 in the cluster consisting of primitive bipotent colony-forming cells and differentiated myoepithelial cells, while expression of NOTCH3 was higher in the committed luminal progenitor cells [22]. Among the NOTCH receptors, breast cancer stem cell activity was dependent significantly on NOTCH4 [36]. In addition, NOTCH signaling has been reported to mediate the tumor resistance to BEV in xenografts of glioblastoma [37]. In the current study, the expression levels of NOTCH4 gradually increased with BEV therapy and this increase was statistically significant in the LT therapy of MV165 group, while the NOTCH3 expression remained similar in response to the ST and LT treatment of BEV. Based on this information and our microarray data, the current study supports the evidence that upregulation of FST and NOTCH4 may form the basis of an environment with cancer stem cell characteristics and confer resistance to anti-VEGF therapy in MV165 xenograft model. Although less dramatic, FST and NOTCH4 levels are also increased in non-VEGF model. Therefore, targeting these pathways may present an important mechanism to prevent resistance to BEV. Further detailed studies will reveal the exact nature of the interaction between FST/BMP/TGFB and NOTCH signaling. However, the balance between BMP/hedgehog and WNT/FGF/NOTCH networks is important for the maintenance of homeostasis among stem and progenitor cells [38, 39]
In order to get a more detailed perspective of the overall changes in gene expression, we analyzed the gene expression data using previously described metagenes. Comparison of gene profiling with metagenes revealed that BEV decreased proliferation, stromal, T cell, and VEGF-related genes in these metagenes, but upregulated the genes associated with myoepithelial/basal phenotype. However, BEV was not effective in decreasing the tumor growth in non-VEGF ML20 model and upregulated most of the genes, in particular metagenes related to the proliferation and tumor microenvironment (stromal, T cell, and VEGF metagenes), suggesting the importance of VEGF expression in altered response to BEV therapy, at least in the ST treatment.
Hypoxic adaptation to BEV treatment is also observed in other experimental models [40]. Our data indicates that increased hypoxia is not a universal response to antiangiogenic treatment and this may dictate the choice of synergistic agents. However, we observed an increase in markers (FST and NOTCH4) implicated with cancer stem cells. This finding is collaborated by a recent study which emphasized that anti-angiogenic agents increase breast cancer stem cells. Although these authors attribute the cancer stem cell phenotype to tumor hypoxia [41], as indicated in our study, other pathways may be involved.
An important observation of our study was that BEV treatment of MV165 cells resulted in a change in the intrinsic subtype of tumors. BEV treatment resulted in a shift of expression of the “intrinsic” genes (PAM50) within the tumor cells resulting in expression of genes associated with myoepithelial/ basal-like subtype. In congruence with this, there was downregulation of genes associated with luminal subtypes including ER. Retinoic acid receptor [42] pathway, which is associated with luminal differentiation was downregulated in MV165 after BEV treatment. Although markers of basal-like subtype are the subject of an ongoing debate, marker panels have been proposed to include genes that are characteristic of basal epithelial cells which include downregulation of ER and upregulation of one or more high molecular weight/basal keratins (KRT5/6, KRT14, and KRT17) and CDH3 (P-cadherin) [43]. We were able to confirm these intrinsic subtype changes using qRT-PCR methods, whereas KRT14 expression was confirmed using immunohistochemistry as well. Our findings show that BEV treatment has the ability to switch the cells to a more aggressive basal-like phenotype. It is possible that this may eventually alter the response to BEV. Hence, these data may shed light on the transitory effect of BEV observed in the E2100 first-line metastatic breast cancer trial [10], where anti-VEGF-targeted therapy prolonged progression-free survival for a ST in metastatic breast cancer without improving overall survival.
Using mouse specific-genes, we observed that a larger number of genes were significantly altered compared with human-specific genes in both models, supporting the effect of BEV on the pathways regulating the tumor microenvironment. These results confirm the effect of BEV treatment on the tumor microenvironment as reported in an earlier study that analyzed mouse- and human-specific gene expression changes in BEV-resistant xenograft models of human lung adenocarcinoma [44]. In our model, the top mouse genes involved in acquired resistance in MV165 model (SLPI, CCL21, CCL5 (RANTES), and CXCR6) represent pathways associated with tumor angiogenesis, innate and adaptive immune system regulation and macrophage signaling. SLPI is overexpressed in inflammatory breast cancer and has been shown to contribute to induction of invasion-independent metastasis in mice mammary tumors [45, 46]. Chemokines are well known to impact the tumor progression, either by promoting or inhibiting tumor growth and metastasis [47, 48]. CCL5 has been shown to play a role in breast metastatic processes by increasing migration- and invasion-related properties [49]. CXCR6 and its ligand CXCL16 also contribute to the regulation of metastasis and invasion in cancer [50]. Both the proangiogenic signature and the mesenchymal phenotype have also been reported in recent studies using BEV-resistant preclinical models in head and neck squamous cell carcinoma and in glioblastoma, respectively [51, 52]. In conclusion, this study reports the multifaceted response to BEV in experimental tumors depending on the levels of VEGF expression. These include upregulation of cellular pathways related to resistance as well as a change in the “intrinsic” subtype and acquisition of cancer stem cell characteristics. The genomic analyses identify several candidate genes/pathways that could potentiate the actions of VEGF and prevent development of resistance. The findings might shed some light on why patients treated with BEV may show partial response as demonstrated by increased progression-free survival but do not have a better overall survival. Furthermore, our data underline stratification of patients based on VEGF expression and provide preclinical evidence to develop relevant druggable targets for combination regimens to anti-VEGF therapy in the clinic.
Supplementary Material
Additional File 1Supplementary Figures and Tables.
Acknowledgements
The authors thank Dr. Kern for kindly donating the ML20 and MV165 cell lines. They also thank Genentech for providing murine anti-VEGF monoclonal antibody.
This work was supported by Breast Cancer Research Foundation grant to George W. Sledge, Jr.
Conflict of Interest
The authors do not have any conflict of interest to disclose.
References
1. Guidi AJ, Schnitt SJ, Fischer L, Tognazzi K, Harris JR, Dvorak HF. et al. Vascular permeability factor (vascular endothelial growth factor) expression and angiogenesis in patients with ductal carcinoma in situ of the breast. Cancer. 1997;80:1945-53
2. Dales JP, Garcia S, Bonnier P, Duffaud F, Carpentier S, Djemli A. et al. [Prognostic significance of VEGF receptors, VEGFR-1 (Flt-1) and VEGFR-2 (KDR/Flk-1) in breast carcinoma]. Annales de pathologie. 2003;23:297-305
3. Relf M, LeJeune S, Scott PA, Fox S, Smith K, Leek R. et al. Expression of the angiogenic factors vascular endothelial cell growth factor, acidic and basic fibroblast growth factor, tumor growth factor beta-1, platelet-derived endothelial cell growth factor, placenta growth factor, and pleiotrophin in human primary breast cancer and its relation to angiogenesis. Cancer research. 1997;57:963-9
4. Konecny GE, Meng YG, Untch M, Wang HJ, Bauerfeind I, Epstein M. et al. Association between HER-2/neu and vascular endothelial growth factor expression predicts clinical outcome in primary breast cancer patients. Clinical cancer research: an official journal of the American Association for Cancer Research. 2004;10:1706-16
5. Schneider BP, Gray RJ, Radovich M, Shen F, Vance G, Li L. et al. Prognostic and Predictive Value of Tumor Vascular Endothelial Growth Factor Gene Amplification in Metastatic Breast Cancer Treated with Paclitaxel with and without Bevacizumab; Results from ECOG 2100 Trial. Clinical cancer research: an official journal of the American Association for Cancer Research. 2013;19:1281-9 doi:10.1158/1078-0432.CCR-12-3029
6. Gasparini G, Toi M, Gion M, Verderio P, Dittadi R, Hanatani M. et al. Prognostic significance of vascular endothelial growth factor protein in node-negative breast carcinoma. Journal of the National Cancer Institute. 1997;89:139-47
7. Gasparini G. Prognostic value of vascular endothelial growth factor in breast cancer. The oncologist. 2000;5(Suppl 1):37-44
8. Foekens JA, Peters HA, Grebenchtchikov N, Look MP, Meijer-van Gelder ME, Geurts-Moespot A. et al. High tumor levels of vascular endothelial growth factor predict poor response to systemic therapy in advanced breast cancer. Cancer research. 2001;61:5407-14
9. Miller KD, Chap LI, Holmes FA, Cobleigh MA, Marcom PK, Fehrenbacher L. et al. Randomized phase III trial of capecitabine compared with bevacizumab plus capecitabine in patients with previously treated metastatic breast cancer. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2005;23:792-9 doi:10.1200/JCO.2005.05.098
10. Miller K, Wang M, Gralow J, Dickler M, Cobleigh M, Perez EA. et al. Paclitaxel plus bevacizumab versus paclitaxel alone for metastatic breast cancer. The New England journal of medicine. 2007;357:2666-76 doi:10.1056/NEJMoa072113
11. Schneider BP, Wang M, Radovich M, Sledge GW, Badve S, Thor A. et al. Association of vascular endothelial growth factor and vascular endothelial growth factor receptor-2 genetic polymorphisms with outcome in a trial of paclitaxel compared with paclitaxel plus bevacizumab in advanced breast cancer: ECOG 2100. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2008;26:4672-8 doi:10.1200/JCO.2008.16.1612
12. Pivot X, Schneeweiss A, Verma S, Thomssen C, Passos-Coelho JL, Benedetti G. et al. Efficacy and safety of bevacizumab in combination with docetaxel for the first-line treatment of elderly patients with locally recurrent or metastatic breast cancer: results from AVADO. European journal of cancer. 2011;47:2387-95 doi:10.1016/j.ejca.2011.06.018
13. Robert NJ, Dieras V, Glaspy J, Brufsky AM, Bondarenko I, Lipatov ON. et al. RIBBON-1: randomized, double-blind, placebo-controlled, phase III trial of chemotherapy with or without bevacizumab for first-line treatment of human epidermal growth factor receptor 2-negative, locally recurrent or metastatic breast cancer. J Clin Oncol. 2011;29:1252-60 doi:10.1200/JCO.2010.28.0982
14. Ebos JM, Lee CR, Christensen JG, Mutsaers AJ, Kerbel RS. Multiple circulating proangiogenic factors induced by sunitinib malate are tumor-independent and correlate with antitumor efficacy. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:17069-74 doi:10.1073/pnas.0708148104
15. Paez-Ribes M, Allen E, Hudock J, Takeda T, Okuyama H, Vinals F. et al. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell. 2009;15:220-31 doi:10.1016/j.ccr.2009.01.027
16. McLeskey SW, Tobias CA, Vezza PR, Filie AC, Kern FG, Hanfelt J. Tumor growth of FGF or VEGF transfected MCF-7 breast carcinoma cells correlates with density of specific microvessels independent of the transfected angiogenic factor. The American journal of pathology. 1998;153:1993-2006 doi:10.1016/S0002-9440(10)65713-6
17. Gokmen-Polar Y, Liu Y, Toroni RA, Sanders KL, Mehta R, Badve S. et al. Investigational drug MLN0128, a novel TORC1/2 inhibitor, demonstrates potent oral antitumor activity in human breast cancer xenograft models. Breast Cancer Res Treat. 2012;136:673-82 doi:10.1007/s10549-012-2298-8
18. Gokmen-Polar Y, Toroni RA, Hocevar BA, Badve S, Zhao Q, Shen C. et al. Dual targeting of EphA2 and ER restores tamoxifen sensitivity in ER/EphA2-positive breast cancer. Breast cancer research and treatment. 2011;127:375-84 doi:10.1007/s10549-010-1004-y
19. Abd El-Rehim DM, Pinder SE, Paish CE, Bell J, Blamey RW, Robertson JF. et al. Expression of luminal and basal cytokeratins in human breast carcinoma. J Pathol. 2004;203:661-71 doi:10.1002/path.1559
20. Cheang MC, Chia SK, Voduc D, Gao D, Leung S, Snider J. et al. Ki67 index, HER2 status, and prognosis of patients with luminal B breast cancer. J Natl Cancer Inst. 2009;101:736-50 doi:10.1093/jnci/djp082
21. Cheang MC, Voduc D, Bajdik C, Leung S, McKinney S, Chia SK. et al. Basal-like breast cancer defined by five biomarkers has superior prognostic value than triple-negative phenotype. Clin Cancer Res. 2008;14:1368-76 doi:10.1158/1078-0432.CCR-07-1658
22. Raouf A, Zhao Y, To K, Stingl J, Delaney A, Barbara M. et al. Transcriptome analysis of the normal human mammary cell commitment and differentiation process. Cell stem cell. 2008;3:109-18 doi:10.1016/j.stem.2008.05.018
23. Karn T, Pusztai L, Holtrich U, Iwamoto T, Shiang CY, Schmidt M. et al. Homogeneous datasets of triple negative breast cancers enable the identification of novel prognostic and predictive signatures. PloS one. 2011;6:e28403. doi:10.1371/journal.pone.0028403
24. Buffa FM, Harris AL, West CM, Miller CJ. Large meta-analysis of multiple cancers reveals a common, compact and highly prognostic hypoxia metagene. British journal of cancer. 2010;102:428-35 doi:10.1038/sj.bjc.6605450
25. Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA. et al. Molecular portraits of human breast tumours. Nature. 2000;406:747-52
26. Perou CM, Jeffrey SS, van de Rijn M, Rees CA, Eisen MB, Ross DT. et al. Distinctive gene expression patterns in human mammary epithelial cells and breast cancers. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:9212-7
27. Sorlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H. et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:10869-74 doi:10.1073/pnas.191367098
28. Parker JS, Mullins M, Cheang MC, Leung S, Voduc D, Vickery T. et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J Clin Oncol. 2009;27:1160-7 doi:10.1200/JCO.2008.18.1370
29. Dowsett M, Nielsen TO, A'Hern R, Bartlett J, Coombes RC, Cuzick J. et al. Assessment of Ki67 in breast cancer: recommendations from the International Ki67 in Breast Cancer working group. J Natl Cancer Inst. 2011;103:1656-64 doi:10.1093/jnci/djr393
30. Sledge GW, Miller KD, Schneider BP, Sweeney CJ. Resistance to antiangiogenesis agents. In: (ed.) Teicher BA. Cancer Drug Resistance. Totowa, New Jersey: Humana Press. 2006:391-410
31. Qu Z, Van Ginkel S, Roy AM, Westbrook L, Nasrin M, Maxuitenko Y. et al. Vascular endothelial growth factor reduces tamoxifen efficacy and promotes metastatic colonization and desmoplasia in breast tumors. Cancer research. 2008;68:6232-40 doi:10.1158/0008-5472.CAN-07-5654
32. Abe Y, Minegishi T, Leung PC. Activin receptor signaling. Growth factors. 2004;22:105-10
33. Welt CK. The physiology and pathophysiology of inhibin, activin and follistatin in female reproduction. Current opinion in obstetrics & gynecology. 2002;14:317-23
34. Sulyok S, Wankell M, Alzheimer C, Werner S. Activin: an important regulator of wound repair, fibrosis, and neuroprotection. Molecular and cellular endocrinology. 2004;225:127-32 doi:10.1016/j.mce.2004.07.011
35. Krneta J, Kroll J, Alves F, Prahst C, Sananbenesi F, Dullin C. et al. Dissociation of angiogenesis and tumorigenesis in follistatin- and activin-expressing tumors. Cancer research. 2006;66:5686-95 doi:10.1158/0008-5472.CAN-05-3821
36. Harrison H, Farnie G, Howell SJ, Rock RE, Stylianou S, Brennan KR. et al. Regulation of breast cancer stem cell activity by signaling through the Notch4 receptor. Cancer research. 2010;70:709-18 doi:10.1158/0008-5472.CAN-09-1681
37. Li JL, Sainson RC, Oon CE, Turley H, Leek R, Sheldon H. et al. DLL4-Notch signaling mediates tumor resistance to anti-VEGF therapy in vivo. Cancer research. 2011;71:6073-83 doi:10.1158/0008-5472.CAN-11-1704
38. Katoh M. Networking of WNT, FGF, Notch, BMP, and Hedgehog signaling pathways during carcinogenesis. Stem cell reviews. 2007;3:30-8
39. Guo S, Liu M, Gonzalez-Perez RR. Role of Notch and its oncogenic signaling crosstalk in breast cancer. Biochimica et biophysica acta. 2011;1815:197-213 doi:10.1016/j.bbcan.2010.12.002
40. Kumar K, Wigfield S, Gee HE, Devlin CM, Singleton D, Li JL. et al. Dichloroacetate reverses the hypoxic adaptation to bevacizumab and enhances its antitumor effects in mouse xenografts. Journal of molecular medicine. 2013;91:749-58 doi:10.1007/s00109-013-0996-2
41. Conley SJ, Gheordunescu E, Kakarala P, Newman B, Korkaya H, Heath AN. et al. Antiangiogenic agents increase breast cancer stem cells via the generation of tumor hypoxia. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:2784-9 doi:10.1073/pnas.1018866109
42. Gokmen-Polar Y. Goswami C CR, Oelschlager KM, Maetzold D, Vladislav IT, Shirar KL, Kesler K, Loehrer PJ Sr, Badve SS. A 19-gene prognostic GEP signature (DecisionDX-Thymoma) to determine metastatic risk associated with thymomas. J Clin Oncol. 2012;30(suppl):abstr7106
43. Badve S, Dabbs DJ, Schnitt SJ, Baehner FL, Decker T, Eusebi V. et al. Basal-like and triple-negative breast cancers: a critical review with an emphasis on the implications for pathologists and oncologists. Modern pathology: an official journal of the United States and Canadian Academy of Pathology, Inc. 2011;24:157-67 doi:10.1038/modpathol.2010.200
44. Cascone T, Herynk MH, Xu L, Du Z, Kadara H, Nilsson MB. et al. Upregulated stromal EGFR and vascular remodeling in mouse xenograft models of angiogenesis inhibitor-resistant human lung adenocarcinoma. The Journal of clinical investigation. 2011;121:1313-28 doi:10.1172/JCI42405
45. Bertucci F, Finetti P, Rougemont J, Charafe-Jauffret E, Nasser V, Loriod B. et al. Gene expression profiling for molecular characterization of inflammatory breast cancer and prediction of response to chemotherapy. Cancer research. 2004;64:8558-65 doi:10.1158/0008-5472.CAN-04-2696
46. Sugino T, Kusakabe T, Hoshi N, Yamaguchi T, Kawaguchi T, Goodison S. et al. An invasion-independent pathway of blood-borne metastasis: a new murine mammary tumor model. The American journal of pathology. 2002;160:1973-80 doi:10.1016/S0002-9440(10)61147-9
47. Allavena P, Sica A, Solinas G, Porta C, Mantovani A. The inflammatory micro-environment in tumor progression: the role of tumor-associated macrophages. Critical reviews in oncology/hematology. 2008;66:1-9 doi:10.1016/j.critrevonc.2007.07.004
48. Ben-Baruch A. The multifaceted roles of chemokines in malignancy. Cancer metastasis reviews. 2006;25:357-71 doi:10.1007/s10555-006-9003-5
49. Soria G, Ben-Baruch A. The inflammatory chemokines CCL2 and CCL5 in breast cancer. Cancer letters. 2008;267:271-85 doi:10.1016/j.canlet.2008.03.018
50. Deng L, Chen N, Li Y, Zheng H, Lei Q. CXCR6/CXCL16 functions as a regulator in metastasis and progression of cancer. Biochimica et biophysica acta. 2010;1806:42-9 doi:10.1016/j.bbcan.2010.01.004
51. Gyanchandani R, Ortega Alves MV, Myers JN, Kim S. A Proangiogenic Signature Is Revealed in FGF-Mediated Bevacizumab-Resistant Head and Neck Squamous Cell Carcinoma. Molecular cancer research: MCR. 2013;11:1585-96 doi:10.1158/1541-7786.MCR-13-0358
52. Piao Y, Liang J, Holmes L, Henry V, Sulman E, de Groot JF. Acquired resistance to anti-VEGF therapy in glioblastoma is associated with a mesenchymal transition. Clin Cancer Res. 2013;19:4392-403 doi:10.1158/1078-0432.CCR-12-1557
Author contact
![]() Corresponding authors: Yesim Gökmen-Polar, PhD. Indiana University School of Medicine, Department of Pathology & Laboratory Medicine, 635 Barnhill Drive, MS 128, Indianapolis, IN 46202-5120. E-mail: ypolaredu Or George W. Sledge, Jr. M.D. Stanford School of Medicine, Department of Medicine, Division of Oncology, 269 Campus Drive, Stanford CA 94305-5151. E-mail: gsledgeedu.
Corresponding authors: Yesim Gökmen-Polar, PhD. Indiana University School of Medicine, Department of Pathology & Laboratory Medicine, 635 Barnhill Drive, MS 128, Indianapolis, IN 46202-5120. E-mail: ypolaredu Or George W. Sledge, Jr. M.D. Stanford School of Medicine, Department of Medicine, Division of Oncology, 269 Campus Drive, Stanford CA 94305-5151. E-mail: gsledgeedu.
Citation styles
APA
Gökmen-Polar, Y., Goswami, C.P., Toroni, R.A., Sanders, K.L., Mehta, R., Sirimalle, U., Tanasa, B., Shen, C., Li, L., Ivan, M., Badve, S., Sledge Jr, G.W. (2014). Gene Expression Analysis Reveals Distinct Pathways of Resistance to Bevacizumab in Xenograft Models of Human ER-Positive Breast Cancer. Journal of Cancer, 5(8), 633-645. https://doi.org/10.7150/jca.8466.
ACS
Gökmen-Polar, Y.; Goswami, C.P.; Toroni, R.A.; Sanders, K.L.; Mehta, R.; Sirimalle, U.; Tanasa, B.; Shen, C.; Li, L.; Ivan, M.; Badve, S.; Sledge Jr, G.W. Gene Expression Analysis Reveals Distinct Pathways of Resistance to Bevacizumab in Xenograft Models of Human ER-Positive Breast Cancer. J. Cancer 2014, 5 (8), 633-645. DOI: 10.7150/jca.8466.
NLM
Gökmen-Polar Y, Goswami CP, Toroni RA, Sanders KL, Mehta R, Sirimalle U, Tanasa B, Shen C, Li L, Ivan M, Badve S, Sledge Jr GW. Gene Expression Analysis Reveals Distinct Pathways of Resistance to Bevacizumab in Xenograft Models of Human ER-Positive Breast Cancer. J Cancer 2014; 5(8):633-645. doi:10.7150/jca.8466. https://www.jcancer.org/v05p0633.htm
CSE
Gökmen-Polar Y, Goswami CP, Toroni RA, Sanders KL, Mehta R, Sirimalle U, Tanasa B, Shen C, Li L, Ivan M, Badve S, Sledge Jr GW. 2014. Gene Expression Analysis Reveals Distinct Pathways of Resistance to Bevacizumab in Xenograft Models of Human ER-Positive Breast Cancer. J Cancer. 5(8):633-645.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) License. See http://ivyspring.com/terms for full terms and conditions.