Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2014; 5(2):133-142. doi:10.7150/jca.7773 This issue Cite
Research Paper
Preclinical Development of ONC1-13B, Novel Antiandrogen for Prostate Cancer Treatment
Alexandre V. Ivachtchenko1,2,3 ![]() , Oleg D. Mitkin2, Elizaveta V. Kudan2, Alexey A. Rjahovsky2, Anton A. Vorobiev2, Andrey S. Trifelenkov2, Natalia A. Shevkun2, Oxana V. Proskurina2, Dmitry V. Kravchenko2, Ruben N. Karapetian2
, Oleg D. Mitkin2, Elizaveta V. Kudan2, Alexey A. Rjahovsky2, Anton A. Vorobiev2, Andrey S. Trifelenkov2, Natalia A. Shevkun2, Oxana V. Proskurina2, Dmitry V. Kravchenko2, Ruben N. Karapetian2
1. ChemDiv Inc., 6605 Nancy Ridge Drive, San Diego, CA 92121, USA
2. Chemical Diversity Research Institute, 141401 Khimki, Rabochaya str, 2a-1, Moscow Reg, Russia
3. Alla Chem LLC., 1835 E. Hallandale Beach Blvd, #442, Hallandale Beach, FL 33009, USA
Received 2013-9-28; Accepted 2013-12-9; Published 2014-1-21
Abstract
Recently new drugs targeting androgen-dependent axis have been approved for the treatment of castration-resistant prostate cancer (CRPC) - Zytiga and Xtandi (formerly MDV3100), several other candidates (for example, ARN-509) are in early phases of clinical trials. However despite significant improvement in overall survival with new treatments it is evident that resistance to these drugs develops. One of the approaches to overcome it is combination therapy and from this point of view some potential for drug-drug interactions can limit the application of the drug.
We describe here the preclinical development of ONC1-13B, antagonist of androgen receptor, with similar to MDV3100 and ARN-509 mechanism of action. It efficiently inhibits DHT-stimulated PSA expression and proliferation of prostate cancer cells, prevents binding of androgens to the AR ligand-binding domain, androgen-stimulated AR nuclear translocation and coactivator complex formation. In the LnCaP-Z2 xenograft model of prostate cancer ONC1-13B inhibits tumor growth and suppresses PSA expression. The in vivo activity of ONC1-13B is comparable to that of MDV3100 at similar doses, and even higher, calculated per unit of concentration in plasma. Distribution of ONC1-13B to the brain is less than that shown for MDV3100 and ARN-509, decreasing the risk of GABA-related seizure development. Additionally ONC1-13B induces significantly lower in vitro CYP3A activity than for example MDV3100 (known strong CYP3A inducer) or ARN-509 and could be well suited for co-therapy with drugs that are known CYP3A substrates. Thus ONC1-13B is a new promising antiandrogen demonstrating high efficacy in a preclinical model of prostate cancer, with lower potential for seizures and drug-drug interaction.
Keywords: ONC1-13B, Antiandrogen, Prostate Cancer
Introduction
Prostate cancer (PC) is the second-leading cause of cancer-related death in men in the US, and the sixth most common cause of cancer death worldwide [1]. The majority of prostate cancers is initially androgen dependent but is able to regenerate and survive without the presence of high levels of testosterone. It is known as hormone-resistant or castration-resistant prostate cancer (CRPC) and is associated with elevated androgen receptor (AR) expression, AR mutation or AR gene amplification [2]. The prognosis for patients with prostate cancer varies greatly and is highly dependent on a number of factors, including the stage of the cancer at diagnosis, race, age, and overall health [3]. As a global trend, the prognosis worsens with age, since the mortality rate increases with age at diagnosis. If prostate cancer is diagnosed and treated early it can often be cured by localized therapies like radical prostatectomy and radiotherapy. The more advanced the cancer at diagnosis, the poorer the prognosis: for example, in the US, the five-year survival rate for patients diagnosed with localized prostate cancer is 100%, while the five-year survival rate for patients diagnosed with distant metastases drops to 31% [4].
The most common types of hormone therapy are Gonadotropin Releasing Hormone (GnRH) receptor agonists and antagonists. These are also referred to as Luteinizing-Hormone-Releasing Hormone (LHRH) receptor agonists and antagonists. Another class of hormone therapy is antiandrogen drugs, which competitively bind to the androgen receptor and block the binding of testosterone and DHT. Antiandrogens are often used during the first few weeks of treatment with an LHRH agonist in order to prevent the temporary increase in testosterone levels known as a “testosterone flare,” or “tumor flare,” that occurs. Antiandrogens are also added to LHRH treatment for a Combined Androgen Blockade (CAB), once primary LHRH agonist therapy has failed. However, they are rarely used as monotherapy in the US. AstraZeneca's Casodex and generic bicalutamide are the most popular antiandrogens.
10-20% of prostate cancer patients develop CRPC within approximately 5 years of follow-up. The prevalence of bone metastases present at diagnosis of CRPC is ≥ 84% [5]. Of those patients with no metastases present at diagnosis of CRPC, 33% could expect to develop them within 2 years. Only 37% of patients with CRPC receive chemotherapy, with the remainder receiving only steroids and supportive care. The most common palliative therapies administered to patients with skeletal symptoms were radiotherapy, radionuclide therapy, bisphosphonates and opioids. Progression to CRPC is associated with deterioration in quality of life, and few therapeutic options are currently available to patients with CRPC.
Recently several new drugs have been approved for the treatment of CRPC: Jevtana (cell division inhibitor), Zytiga (CYP17A inhibitor, androgene blockator) and Xtandi (androgen receptor antagonist). Two of them - Zytiga and Xtandi, are targeting androgen-dependant axis, thus providing evidence of significant importance of this pathway as a target for treatment. Another antiandrogen, ARN-509, is currently in a Phase II clinical trial in patients with advanced castration-resistant prostate cancer [6].
We describe here the preclinical development of 4-[(R)-3-(4-Cyano-3-trifluoromethyl-phenyl)-4-oxo-2-thioxo-7-oxa-1,3-diaza-spiro[4.4]non-1-yl]-2-fluoro-N-methyl-benzamide (ONC1-13B), antagonist of androgen receptor, with mechanism of action similar to MDV3100. Its antitumor activity is higher than MDV3100 and ARN-509 in vitro and in vivo, calculated per unit of concentration in plasma. Like other antiandrogens [7] ONC1-13B binds with low affinity to GABA receptors, but taking into account low brain distribution compared to MDV3100 and ARN-509, lower seizure potential is suggested for ONC1-13B. Also we demonstrated here that ONC1-13B is a significantly weaker inducer of CYP3A compared to MDV3100 and ARN-509, consequently has low potential for drug-drug interactions and can be well suited for co-therapy with different drugs.
Materials and Methods
All experimental procedures using commercially available kits were performed according to manufacturer`s instructions.
Chemicals
Bicalutamide (Tocris, cat.#3389), ARN-509 (Selleck Chemicals, cat.#S2840), MDV3100 (Selleck Chemicals, cat.#S1250), DHT (Wako, cat.#045-26071). ONC1-13B was synthesized at Chemical Diversity Research Institute. Stock and serial dilutions were in dimethylsulfoxide (DMSO) unless otherwise specified.
Cell-lines
LNCaP (ATCC, cat.#CRL-1740) were propagated in RPMI-1640 (Invitrogen, cat.#11835-030) + 10% FCS (Hyclone, cat.#SH300070.03) + 1%AAS (Sigma, cat.#5955).
LNCaP-Z2 cells were grown in RPMI 1640, Glutamax 1 (Invitrogen, cat.#61870-010) with 10% FCS, 100 units penicillin/ml, and 100µg of streptomycin/ml.
Ligand binding studies
PolarScreen™ Androgen Receptor Competitor Assay (Invitrogen, cat.#PV4293) was used according to manufacturer`s procedure to determine relative in vitro binding affinity of ONC1-13B and MDV3100 for the rat AR ligand-binding domain (LBD). Each concentration of compounds was tested in duplicates.
LanthaScreen™ TR-FRET Androgen Receptor Coactivator Assay (Invitrogen, cat.#PV4381) was used in agonist and antagonist mode according to manufacturer`s procedure to determine the influence of ONC1-13B, MDV3100 and Bicalutamide on ligand-dependent coactivator reqruitment. Each concentration of compounds was tested in duplicates.
GABA binding experiment was carried out at CEREP, Paris, France. Briefly, membrane homogenates of rat cerebral cortex were incubated for 60 min at RT with 10 nM [3H] GABA in the absence or presence of 10uM ONC1-13B. Nonspecific binding was determined in the presence of 100 μM GABA. Radioactivity was counted in a scintillation counter (Topcount, Packard) using a scintillation cocktail (Microscint 0, Packard). The results were expressed as a percent inhibition of the control radioligand specific binding.
Proliferation assay
Trypsinized LnCAP cells were adjusted to a concentration of 50,000 cells per mL in RPMI-1640 + 10% Charcoal-stripped serum (CSS) (Invitrogen, cat.# 12676-011) +1%AAS and dispensed in 200 μL aliquots into 96-well Poly-D-Lysine plate (BD, cat.# 356651). Cells were incubated for 72 hours, after which serial dilutions of tested compounds were added to the final concentration of 1nM-10uM. DHT was added to the final concentration of 1nM. Final concentration of DMSO is 0.1%. Next day 50 ul of medium from each well was used to measure concentration of PSA (Vector-Best, cat.#8458). After 5 days' incubation, CellTiter-Glo Luminescent Cell Viability Assay (Promega, cat.#G7572) was added and Relative Luminescence Units (RLUs) measured.
AR nuclear translocation
Inhibition of androgen-stimulated nuclear translocation of AR by Bicalutamide, MDV3100 and ONC1-13B was tested in DiscoverX using PathHunter® NHR NT.
In vivo pharmacology
All animal studies were carried out under protocols approved by the Institutional Animal Care and Use Committees and institutional guidelines for the proper, humane use of animals in research were followed.
In vivo xenograft experiment to determine anti-tumor response was carried out in male CB17-SCID mice (Charles River GmbH). On day 0, the appropriate amount of 2x106 LNCaP-Z2 tumor cells in 200µl PBS:Matrigel 1:1 were implanted into the subcutaneous space of the left flank of all mice. On day 18, after a mean tumor volume of approx. 160-190 mm3 was reached, all tumor-bearing animals were randomized into 6 study groups according to tumor sizes. On the following day (day 19), treatment was initiated in all groups and lasted until day 40. All compounds were administered daily by oral gavage. ONC1-13B, MDV3100 and bicalutamide drug stocks were prepared in 0.5% Methylcellulose. At necropsy, 24 hours after the last drug administration (or 16 hours in case of the double treatment), blood samples were taken from all animals with microcapillaries via retroorbital vein puncture, after animals were anaesthetised with isoflurane. To obtain EDTA-plasma, blood samples were directly collected into EDTA-tubes and centrifuged for 10 min at 5000g and 4°C. Plasma was used to measure concentration of PSA (Quantikine; distributed by R&D Systems Inc.; Cat.#DKK300) and concentration of tested compounds. Statistical analyses were performed using GraphPad Prism.
In vivo pharmacodynamic studies
ONC1-13B formulated in 0.5% Methylcellulose was administered daily for 14 days at 0, 30, 100 and 300 mg/kg by oral gavage (po) to male rats. The vehicle group received only 0.5% Methylcellulose. On Day 15, the animals were weighed, sacrificed and necropsied, organ weight was recorded.
Rat and dog pharmacokinetics
Male Sprague Dawley rats (250-350g) obtained from the Animal Breeding Facility, Branch of Shemyakin&Ovcinnikov Insitute of Bioorganic Chemistry RAS, Moscow Region and male beagle dogs (10-12 kg) obtained from the Animal Breeding Facility, Russian Research Center of Biologically Active Substances, Moscow region were used for pharmacokinetics studies.
Plasma samples (45 μL) were mixed with 5 μl of acetonitrile:H2O (1:1 v/v), then precipitated with 150 μl of acetonitrile containing internal standard 50 ng/ml tolbutamide. Samples were incubated at -20°C for 15 min followed by centrifugation at 11 000g for 10 min, 130 uL of supernatants were transferred to new plate for LC/MS/MS analysis. Calibrations and quality control samples were prepared in 45 uL of intact plasma spiked with 5 uL of 10x standards in acetonitrile:H2O (1:1 v/v) in the range of 0,001 - 10 ug/ml.
Tumor, prostate and brain tissue-distribution
Pre-weighed tissue samples were homogenized using Omni MultiMix 200 system (Omni Corp., USA) in 3 (tumor) or 5 (prostate, brain) volumes of deionized water (w/v) and processed for 45 s at 4000 rpm followed by 30 s centrifugation at 30 g. Tumor homogenate (90 μL) was combined with 10 μL of acetonitrile:water, 1:1, v/v, then 500 uL of ice-cold acetonitrile containing 50 ng/ml of internal standard tolbutamide were added. Prostate and brain homogenate (135 μL) was combined with 15 μL of acetonitrile:water, 1:1, v/v, then 1650 uL of ice-cold acetonitrile containing 50 ng/ml of internal standard tolbutamide were added.
Samples were incubated on ice for 30 min, precipitated proteins were removed by centrifugation at 10000 g for 15 min. Supernatants were used for LC-MS/MS analysis of compound's content.
Calibration curves were prepared using intact tissue homogenates and 10x standard solutions in acetonitrile:water, 1:1 according to the procedures described above.
LC-MS/MS method
The LCMS/MS analysis was performed with Agilent 1290 UPLC coupled with Qtrap5500 mass-spec (ABSciex,USA). Data acquisition was done with Analyst 1.5.2 software. 1 uL of samples were injected onto YMC-Ultra HT Hydrosphere C18 column (50х2 mm,2µm,12nm) with H2O: MeCN(30:70) +0.1% formic acid mobil phase under isocratic conditions with 400 uL/min flow rate. Positive-ion multiple reaction monitoring was used for the MS/MS detection of ONC1-13B, MDV3100 and negative for Bicatulamide. Entrance, declustering and exit potentials, collision energy and other specific MS/MS parameters were optimized for each compound. Mass transitions m/z were: ONC1-13B - 493.1/380.0, MDV3100 - 265.2/209.0 and 429.0/255.0 for Bicatulamide.
Pharmacokinetic parameters were calculated using non-compartmental analysis provided by the software WinNonlin Professional 5.2 (Pharsight Corporation).
In vitro plasma-protein binding
Free (unbound) fractions of ONC1-13B or MDV3100 in plasma were determined in vitro by equilibrium dialysis (Pierce, cat.# 89810, 89811) at 1 uM in mouse, rat, dog and human plasma. The assay was performed in plasma diluted with PBS pH=7.2 (1:1). Equilibrium between plasma and buffer chambers divided by membrane with 8 kDa cut-off were reached during 4h incubation at 37˚C and gentle shaking.
CYP3A induction
CYP3A induction experiment was carried out at CEREP, Redmond, USA. Briefly, tested compounds were incubated with human plateable cryopreserved hepatocytes (0.7 mln viable cells/ml) at 1 and 10 uM for 48 hours. Midazolam (CYP3A substrate) was added to final concentration of 10 uM and incubated for 30 min. Concentration of 1-hydroxymidazolam was measured by LCMSMS. Peak areas corresponding to the 1-hydroxymidazolam were recorded. The fold induction was then calculated by comparing peak area obtained in the test compound treated cells to that obtained in the negative control (solvent treated cells). The percent of positive control was calculated by comparing the peak area obtained in the test compound treated cells to that obtained in the positive control (known CYP inducer treated cells), as described below:
%Positive control=(activity of test drug treated cells-activity of negative control)*100%/(activity of positive control-activity of negative control).
Results
ONC1-13B binds AR, impairs nuclear localization of AR and inhibits androgen-dependent gene expression
ONC1-13B (Fig. 1A) is a synthetic nonsteroidal antiandrogen that fully inhibits androgen-dependent gene expression. It was identified by its ability to inhibit AR-dependent PSA expression in prostate cancer cells LnCAP. Cells were cultured in medium with 5% CSS for 3 days and then treated with tested compounds in the presence of 5-α-dihydrotestosterone (DHT). Expression of PSA was measured in culture medium 24 hours thereafter (Fig. 1B). ONC1-13B inhibits DHT-induced PSA expression (Ki 20nM) around 10-fold more efficiently than bicalutamide (Ki 190 nM, data not shown) and slightly more efficiently than two other clinical stage antiandrogens, MDV3100 (Ki 30.8 nM) and ARN-509 (Ki 38.4 nM). LnCAP cells were cultured further for 5 days and the number of viable cells was calculated. As it is shown on the Fig. 1C, ONC1-13B like others antiandrogens inhibits DHT-induced cell proliferation.
ONC1-0013B inhibits AR activity in vitro. A. ONC1-0013B structure. B. LnCAP cells cultured (10% CSS) for 3 days, then treated with tested compounds in presence of 1nM DHT for 1 day. PSA expression plotted as percentage of vehicle control (DMSO; n=2, mean±SEM). Ki values: 20.0±5.5nM (ONC1-13B), 30.8±7.7nM (MDV3100), 38.4nM (ARN-509). Mean±SEM from 5 replicate experiments (except ARN-509). C. LnCAP cells cultured (10% CSS) for 3 days, then treated with tested compounds in presence of 1nM DHT for 5 days. Viable cells plotted as percentage of vehicle control (DMSO; n=2, mean±SEM). IC50 values: 30nM (ONC1-13B), 148nM (MDV3100), 240nM (ARN-509). D. Competitive-binding assay vs AR ligand Fluormone™ (PolarScreen™ Androgen Receptor Competitor Assay). IC50 values: 19nM (DHT), 7.9uM (ONC1-13B), 16.3uM (MDV3100).
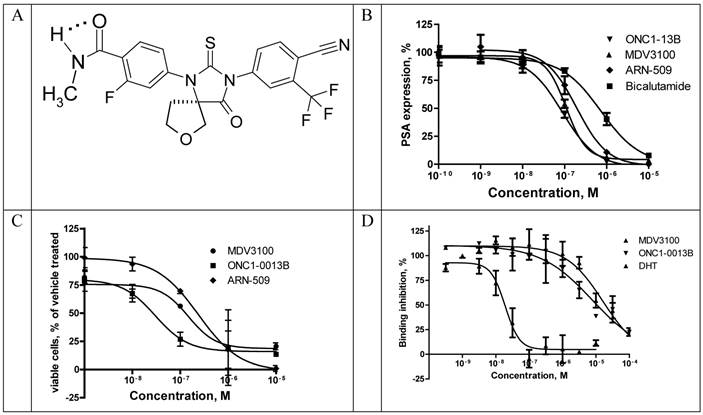
Binding to rat AR ligand-binding domain was measured in competition with fluorescently labeled AR ligand Fluormone™ using commercial assay kit and fluorescence polarization as a read-out. ONC1-13B binds to AR with the same efficacy as MDV3100 (Fig. 1D).
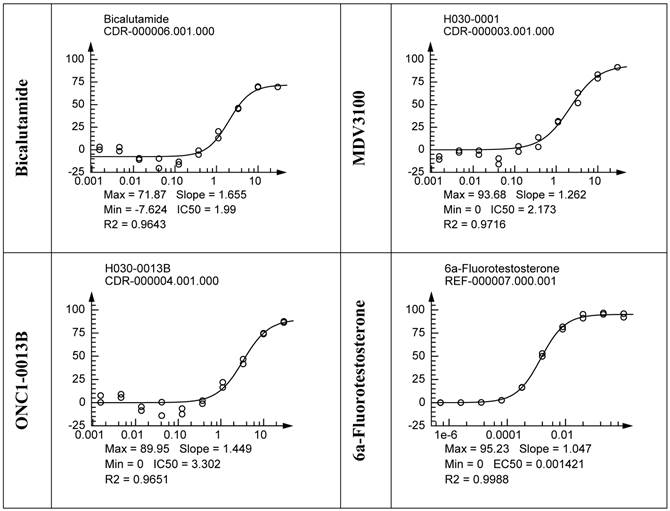
Translocation of AR from cytoplasm to the nucleus upon ligand binding is a highly regulated essential step in AR-mediated gene expression. To determine whether ONC1-13B affect AR nuclear localization commercial assay based on the Enzyme Fragment Complementation was used. Translocation of AR tagged with ProLabelTM to the nucleus generates active beta-Galactosidase and produces chemiluminescent signal. Results are shown in the Fig. 2. Bicalutamide doesn`t completely inhibit AR nuclear translocation as it is partial agonist. ONC1-13B and MDV3100 completely inhibit 6a-Fluotestosterone induced AR nuclear translocation with similar efficacy.
PathHunter® NHR cells (DiscoverX) were pre-incubated with tested compounds followed by addition of 6α-Fluorotestosterone at 7.5nM (EC80). Percentage of androgen-stimulated nuclear translocation of AR inhibition was normalized to the maximal and minimal response observed in the presence of 6α-Fluorotestosterone and vehicle. IC50 values: 3.3uM (ONC1-13B), 2.2uM (MDV3100, 2.0uM (Bicalutamide). N=2.
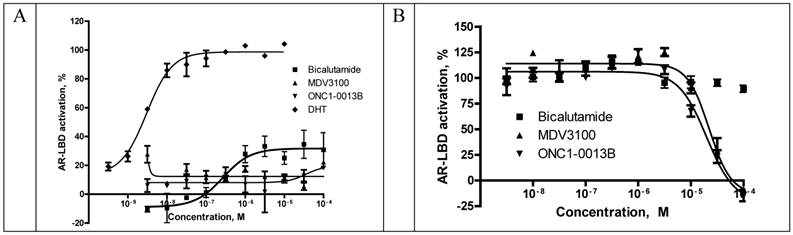
Recruitment of coactivator proteins and formation of transcription complex is another important step for induction of AR-dependent gene expression. Influence of ONC1-13B on ligand-dependent coactivator-recruitment was measured in commercial coactivator assay kit using TR-FRET as a read-out. Compounds were tested in agonist and antagonist modes in competition with DHT. As it is shown on the Fig. 3A ONC1-13B and MDV3100 are significantly more active compared to Bicalutamide and have similar activity in antagonist mode, while in agonist mode (Fig. 3B) only Bicalutamide induces coactivator recruitment. Together with previous results it shows that ONC1-13B is full antagonist of androgen receptor.
Competitive-binding assay vs AR Ligand-Binding Domain (LanthaScreen™ TR-FRET Androgen Receptor Coactivator Assay). A. Agonist mode - AR-LBD is added to test compounds followed by addition of a mixture of the fluorescein-coactivator peptide and terbium anti-GST antibody. EC50 for DHT = 3nM. N=2. B. Antagonist mode- AR-LBD is added to compounds followed by addition of a mixture of EC80 DHT, fluorescein-coactivator peptide, and terbium anti-GST antibody. IC50 values: 19.9uM (ONC1-13B), 22.3uM (MDV3100). N=2.
ONC1-0013B efficiently inhibits tumor growth in the xenograft model of prostate cancer
Antitumor efficacy of ONC1-0013B was tested in xenograft model of prostate cancer. Male immunodeficient mice harboring LnCaP-Z2 tumors were treated orally for 21 days with either vehicle or ONC1-13B at 20 and 50 mg/kg once daily and at 20 mg/kg twice daily. MDV3100 was administered at 10 mg/kg once daily, Bicalutamide at 50 mg/kg once daily. 24 hours after last treatment blood samples were collected from all animals and used for measurement of PSA expression and concentration of drugs. Also concentration of drugs and Ki67 expression was measured in tumor samples.
The antitumor efficacy of ONC1-13B administered once daily at 20 or 50 mg/kg is comparable to MDV3100, administered once daily at 10mg/kg, whereas Bicalutamide turned out to be less effective. ONC1-13B, administered twice daily at 20mg/kg, shows the highest antitumor efficacy. Analysis of individual tumors revealed that MDV3100 and ONC1-13B stimulate tumor regression. In the group treated with ONC1-13B twice daily 4 of 8 tumors regressed (Fig. 4C).
Antitumor activity of ONC1-13B in prostate cancer xenograft model. SCID male mice bearing LnCaP-Z2 tumors (mean volume ~160-190 mm3) treated daily by oral gavage with vehicle or drugs during 21 days. A. Tumor weight by the end of the study. Mean±SEM are presented. Unpaired t-test: vehicle vs MDV3100, P=0.0093, vs Bicalutamide, P=0.0830, vs ONC1-13B 20 mg/kg, P=0.0104, 50 mg/kg, P=0.0148, 20 mg/kg twice a day, P=0.0054. B. PSA expression by the end of the study. Plasma samples were collected 24 hours after last drug administration. Mean±SEM are presented. Unpaired t-test: vehicle vs MDV3100, P=0.0140, vs Bicalutamide, P=0.0830, vs ONC1-13B 20 mg/kg, P=0.0148, 50 mg/kg, P=0.0379, 20 mg/kg twice a day, P=0.0070. C. Individual tumor volume change by the end of the study. Percentage of change in individual tumor volumes compared to the start of treatment. D. Ki-67 immunohistochemistry on tumor tissue resected 24 hours after final dose. Proliferative activity 62.5±2.5% (Vehicle), 40.0±7.0 (ONC1-13B, 20 mg/kg twice daily), n=8. Percentage of proliferative activity calculated relative to human lymph node tissue served as positive control. Unpaired t-test Vehicle vs ONC1-13B P=0.0216.
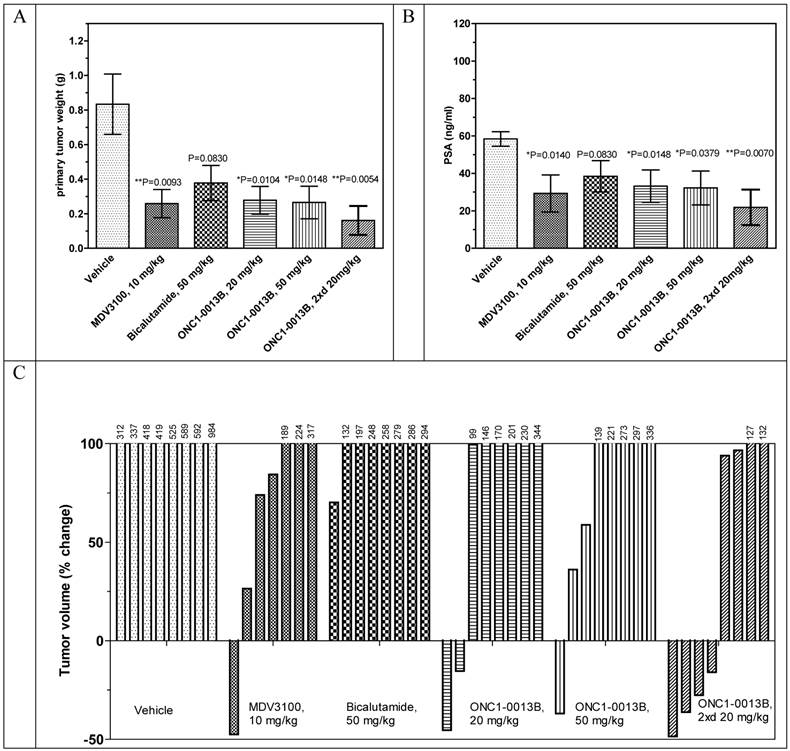
The PSA expression directly correlates with the decreases of tumor sizes (compare Fig. 4B to Figs. 4A). Ki67 staining showed that ONC1-13B inhibits proliferation of prostate cancer cells in vivo (Fig. 5). All together these data show that ONC1-13B is very potent in inhibition of proliferation of prostate cancer cells in vivo. Efficacy of ONC1-13B is comparable or even higher compared to MDV3100 and significantly higher compared to Bicalutamide.
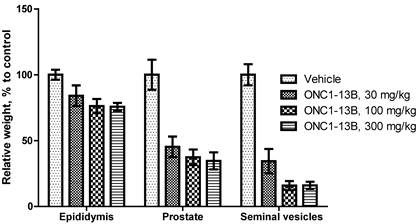
ONC1-13B decreases the weight of epididymis, prostate and seminal vesicles in rats. Male rats treated by oral gavage for 14 days. Organs resected 24 hours after final dose. Relative organ weight plotted as percentage of vehicle control. Mean±SEM (n=6 per group).
To define the relation of efficacy to concentration, we measured plasma and tumor-tissue concentrations of tested drugs following 21 days of continuous dosing. There is linear relationship of the dose of ONC1-13B with its concentration in plasma and tumor after repeated administrations (Table 1). MDV3100 plasma and tumor concentrations are 5- to 10-fold (plasma) and 3- to 9-fold (tumors) higher than for the doses of ONC1-13B with similar activity (Table 1). At the comparable to MDV3100 plasma and tumor concentrations ONC1-13B (20 mg/kg twice daily) demonstrates significantly higher activity. At the same time activity of Bicalutamide is lowest in the study although its concentration and distribution are higher compared to ONC1-13B and MDV3100.
Concentration of tested compounds in plasma and in LnCaP-Z2 xenograft tumor tissue after 21-day dosing period. Mean±SD.
| Group | Concentration in plasma, ng/ml | Concentration in tumor, ng/g | tumor/ plasma |
|---|---|---|---|
| MDV3100, 10 mg/kg | 699.2± 443.2 (n=7) | 426.7± 262.0 (n=6) | 0.53± 0.15 (n=6) |
| Bicalutamide, 50 mg/kg | 3246.3± 1360.9 (n=8) | 8735.3± 8145.6 (n=8) | 2.50± 1.36 (n=8) |
| ONC1-13B, 20 mg/kg | 76.2± 36.8 (n=8) | 52.6± 24.6 (n=6) | 0.78± 0.08 (n=6) |
| ONC1-13B, 2х20 mg/kg | 456.4± 219.1 (n=8) | 442.2± 148.5 (n=7) | 0.72± 0.05 (n=4) |
| ONC1-13B, 50 mg/kg | 154.4± 87.5 (n=8) | 137.5± 58.6 (n=6) | 0.77± 0.13 (n=6) |
NOTE: Tumors with small size do not allow reliable measurement of concentration and are excluded from calculation
Assessment of in vitro free fraction in plasma indicated that ONC1-13B is less protein bound, resulting in an approximately 3- to 6-fold greater free fraction than MDV3100 in mice, rats, dogs and human plasma (Table 2). These data can explain higher activity of ONC1-13B compared to MDV3100 at similar concentrations.
Plasma protein binding for ONC1-0013B and MDV3100
| Bound fraction, % | ||||
|---|---|---|---|---|
| Cmpd ID | Mice | Rats | Dogs | Human |
| MDV3100 | NM | 96.4 | 97.3 | 98.4 |
| ONC1-13B | 90.7 | 89.7 | 89.5 | 89.3 |
NM - not measured
ONC1-13B induces changes in androgen-dependent tissues in rats
Antiandrigenic activity of ONC1-13B was tested in rats by influence on androgen-dependent reproductive organs. ONC1-13B was administered orally at 30, 100 and 300 mg/kg/day to rats during 14 days. As it is shown on the Figure 5, ONC1-13B significantly and dose-dependently decreases the weight of prostate, epididymis and seminal vesicles.
In another experiment it was shown that ONC1-13B significantly decreases concentration of spermatozoon in rats (table 3).
Concentration of spermatozoon in epididymis of male rats. Male rats treated by oral gavage for 60 days. Mean±SEM (n=6 per group).
| Vehicle | ONC1-13В 30 mg/kg | ONC1-13В 300 mg/kg | |
|---|---|---|---|
| Concentration, х 106/ml, M±m | 64,33 ± 3,37 | 29,00 ± 2,73 | 16,00 ± 2,37 |
Pharmacokinetics of ONC1-13B in mice, rats and dogs
ONC1-13B is completely absorbed after oral administration (oral bioavailability in rats 100%, table 4). Concentration of ONC1-13B is ascending in dose-proportional manner after repeated administrations at doses up to 100 mg/kg in rats. In dogs ONC1-13B is slowly absorbed and eliminated - concentration in plasma was growing after single administration that didn`t allow to calculate t1/2. After multiple doses in dogs concentration of ONC1-13B (C24h) was increased twice compared to single administration and reached steady-state after 10 administrations (table 5). These data support once daily oral dosing in human.
Pharmacokinetic parameters for ONC1-0013В after single IV and single and multiple oral doses to rats (n=3).
| Parameter | Units | Single IV | Single PO | Multiple PO, after 14 administrations | ||
|---|---|---|---|---|---|---|
| 1 mg/kg | 5 mg/kg | 30 mg/kg | 100 mg/kg | 300 mg/kg | ||
| Kel | 1/h | 0.167 | 0.13 | 0.19 | 0.09 | 0.11 |
| T1/2 | h | 4.1 | 5.2 | 4 | 8 | 6 |
| Tmax | h | 2 | 4 | 4 | 4 | |
| Cmax | ng/mL | 531 | 1 062 | 7 162 | 16 357 | 20 533 |
| AUCINF_obs | h*ng/mL | 2 572 | 13 105 | 95 000 | 268 000 | 314 000 |
| MRTINF_obs | h | 5.3 | 7 | 4 | 10 | 7 |
| C24h | ng/mL | 51 | 159 | 2 794 | 2 122 | |
| Сl | ml/min/kg | 6.4 | ||||
| Vss_obs | L/kg | 2.0 | ||||
Concentration of ONC1-13В after single and multiple oral doses to dogs at 10 mg/kg (n=4).
| # of dose | time, h | Concentration, ug/ml | SD |
|---|---|---|---|
| 1 | 2 | 1.25 | 0.8 |
| 4 | 1.73 | 0.9 | |
| 6 | 1.8 | 0.9 | |
| 8 | 2.13 | 0.9 | |
| 24 | 3.63 | 1.4 | |
| 10 | 24 | 7.5 | 3.1 |
| 20 | 24 | 11 | 3 |
| 27 | 24 | 7.9 | 2.9 |
| 28 | 2 | 7.03 | 2.4 |
| 4 | 6.98 | 2.5 | |
| 6 | 7.75 | 2.5 | |
| 8 | 6.35 | 1.8 | |
| 24 | 7.03 | 2.7 |
Distribution of ONC1-13B to target tissues (SC implanted tumors- table 1 and prostate - table 6) is higher than MDV3100. Prostate/plasma ratio for ONC1-13B = 1.2-1.6 vs 0.3 for MDV3100 after single IV administration to rats.
Distribution to the brain and prostate after single IV administration in rats (Mean±SD, n=3).
| Time | Drug | plasma, ng/ml | brain, ng/g | Brain/Plasma | Prostate ng/g | Prostate/Plasma |
|---|---|---|---|---|---|---|
| 30 min | ONC1-13B, 2 mg/kg | 746±104 | 145±22 | 0.19±0.02 | 1159±164 | 1.55±0.08 |
| MDV3100, 2 mg/kg | 263±19 | 164±19 | 0.62±0.06 | 102±4 | 0.39±0.03 | |
| ARN-509, 1 mg/kg | 251±29 | 241±21 | 0.96±0.03 | 487±73 | 1.94±0.24 | |
| 120 min | ONC1-13B, 2 mg/kg | 522±57 | 89±10 | 0.17±0.35 | 625±108 | 1.20±0.36 |
| MDV3100, 2 mg/kg | 377±153 | 152±31 | 0.40±0.12 | 124±27 | 0.33±0.16 | |
| ARN-509, 1 mg/kg | 153±10 | 176±31 | 1.15±0.27 | 482±120 | 3.15±0.94 |
Like other antiandrogens ONC1-13B binds weakly to GABA receptors - 42% inhibition at 10 uM. It is comparable to the affinity of MDV3100 and ARN-509 (IC50 = 2.7 and 3.0 uM, respectively, [6]) and thus may potentially cause seizures at high dose. But taking into account lower distribution of ONC1-13B to the brain compared to ARN-509 and MDV3100 (table 6), lower seizure-inducing potential of ONC1-13B is suggested.
One of the potential limitations in co-treatment therapy of MDV3100 can be its ability to induce CYP3A activity. ONC1-13B was tested side-by-side with MDV3100 and ARN-509 in CYP3A induction assay in human hepatocytes. As it is shown in the table 7 ONC1-13B is ~2 fold less potent in CYP3A induction at 10uM compared to MDV3100 and ARN-509 and has no activity at 1uM while MDV3100 and ARN-509 are still active. Also it was shown that ONC1-13B doesn`t inhibit activity of different CYP isoforms (1A2, 2C9, 2C19, 2D6, 3A4) in concentrations up to 10 uM. All together it means that ONC1-13B has lower potential for drug-drug interactions compared to MDV3100 and ARN-509 and can be better suited for combination therapy with different drugs.
CYP 3A induction assay.
| Drug | Concentration, uM | Positive control, % |
|---|---|---|
| MDV3100 | 1 | 10.0±2.0 |
| 10 | 73.3±6.8 | |
| ONC1-0013B | 1 | 0.0±2.0 |
| 10 | 42.0±6.1 | |
| ARN-509 | 1 | 16.3±2.5 |
| 10 | 102.7±13.0 | |
| ONC1-0013B | 1 | 3.3±0.6 |
| 10 | 51.3±3.5 |
NOTES: %Positive control = (activity of test compounds treated cells-activity of negative control)*100/( activity of positive control /activity of negative control)
Discussion
ONC1-13B is a new synthetic androgen receptor antagonist with triple mechanism of action. Recently two other antiandrogens with the same mechanism of action were described in the literature - MDV3100 was approved by FDA for the treatment of castration-resistant prostate cancer and ARN-509 that is currently in phase II clinical development in distinct subsets of prostate cancer.
ONC1-13B was first discovered by its ability to inhibit DHT-stimulated PSA expression in prostate cancer cells LnCAP. It was shown that ONC1-13B was c.a.10-fold more active than bicalutamide and slightly more active than MDV3100 and ARN-509 in this assay. Later it was shown that ONC1-13B prevents binding of androgens to AR, nuclear translocation of AR and transcription activator recruitment thus confirming its triple mechanism of action.
ONC1-13B significantly inhibits tumor growth and causes tumor regression in the prostate cancer xenograft model. Additionally expression of biomarkers, namely PSA and Ki67, was inhibited by ONC1-13B and correlated with the decreases of mice tumor sizes. Efficacy of ONC1-0013B is comparable or even higher than MDV3100 and significantly higher compared to Bicalutamide.
Interestingly that in vivo ONC1-13B is more active per unit of plasma concentration than MDV3100 and bicalutamide. Taking into account comparable to MDV3100 activity in vitro, it can be explained by better distribution of ONC1-13B to the tumor and by higher free fraction of ONC1-13B in the plasma. The target-distribution study revealed that concentration of ONC1-13B in prostate is significantly higher than MDV3100 in rats. Interestingly that distribution to the brain (brain/plasma ratio) for ONC1-13B is significantly lower than that of MDV3100 and ARN-509. Taking together all the data about pharmacokinetics, distribution and efficacy, we expect higher therapeutic index with reduced risk of GABAA-related seizure and other adverse effects for ONC1-13B.
Additionally antiandrogenic activity of ONC1-13B was confirmed by the reduction of epididymis, prostate, seminal vesicles and concentration of spermatozoon after repeated administrations in male rats.
It is known that MDV3100 is a strong inducer of CYP3A activity that potentially restricts its use together with other drugs metabolized via CYP3A. In our study it was shown that ONC1-13B is significantly less potent in CYP3A induction than both MDV3100 and ARN-509. Thus ONC1-13B should be better suited for co-treatment therapy with other drugs, including CYP3A substrates, for example with Zytiga.
Preclinical development of ONC1-13B is currently completed and it is submitted for phase I clinical study in metastatic CPRC patients to determine pharmacokinetics, safety, and pilot efficacy.
Acknowledgements
This study was supported by the Grant from Ministry of Industry and Trade of Russian Federation (11411.1008700.13.0.80).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J. Clin. 2010;60:277-300
2. Scher HI, Sawyers CL. Biology of progressive, castration-resistant prostate cancer: directed therapies targeting the androgen-receptor signaling axis. J Clin Oncol. 2005;23:8253-61
3. Crawford E. Epidemiology of prostate cancer. Urology. 2003;62:3-12
4. Howlader N, Noone AM, Krapcho M, Neyman N, Aminou R, Waldron W, Altekruse SF, Kosary CL, Ruhl J, Tatalovich Z, Cho H, Mariotto A, Eisner MP, Lewis DR, Chen HS, Feuer EJ, Cronin KA. SEER Cancer Statistics Review, 1975-2009 (Vintage 2009 Populations), National Cancer Institute.
5. Kirby M, Hirst C. et al. Characterising the castration-resistant prostate cancer population: a systematic review. Int J Clin Pract. 2011;65(11):1180-1192
6. Clegg NJ, Wongvipat J, Tran C, Ouk S, Dilhas A, Joseph J, Chen Y, Grillot K, Bischoff ED, Cai L, Aparicio A, Dorow S, Arora V, Shao G, Qian J, Zhao H, Yang G, Cao C, Sensintaffar J, Wasielewska T, Herbert MR, Bonnefous C, Darimont B, Scher HI, Smith-Jones PM, Klang M, Smith ND, de Stanchina E, Wu N, Ouerfelli O, Rix P, Heyman R, Jung ME, Sawyers CL, Hager JH. ARN-509: a novel anti-androgen for prostate cancer treatment. Cancer Res. 2012Mar15;72(6):1494-1503
7. Foster WR, Car BD, Shi H, Levesque PC, Obermeier MT, Gan J, Arezzo JC, Powlin SS, Dinchuk JE, Balog A, Salvati ME, Attar RM, Gottardis MM. Drug safety is a barrier to the discovery and development of new androgen receptor antagonists. The Prostate. 2011;71(5):480-8
Author contact
![]() Corresponding author: avcom
Corresponding author: avcom