Journal of Cancer
ISSN: 1837-9664
3.2
Impact Factor
ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2014; 5(2):115-124. doi:10.7150/jca.8084 This issue Cite
Research Paper
An Ultra-Sensitive Immunoassay for Quantifying Biomarkers in Breast Tumor Tissue
Carol B. Fowler1, Yan-Gao Man2, Jeffrey T. Mason1 ![]()
1. Laboratory of Proteomics and Protein Science, Baltimore Veterans Affairs Medical Center, Baltimore, MD, USA;
2. Bon Secours Cancer Institute, Bon Secours Health System, Richmond, VA, USA.
Received 2013-11-8; Accepted 2014-1-1; Published 2014-1-5
Citation:
Fowler CB, Man YG, Mason JT. An Ultra-Sensitive Immunoassay for Quantifying Biomarkers in Breast Tumor Tissue. J Cancer 2014; 5(2):115-124. doi:10.7150/jca.8084. https://www.jcancer.org/v05p0115.htm
Other stylesAbstract
Urokinase-type plasminogen activator (uPA) and plasminogen activator inhibitor type-1 (PAI-1) have been validated at the highest level of evidence as clinical biomarkers of prognosis in breast cancer. The American Society of Clinical Oncology recommends using uPA and PAI-1 levels in breast tumors for deciding whether patients with newly diagnosed node-negative breast cancer can forgo adjuvant chemotherapy. The sole validated method for quantifying uPA and PAI-1 levels in breast tumor tissue is a colorimetric ELISA assay that takes 3 days to complete and requires 100-300 mg of fresh or frozen tissue. In this study we describe a new assay method for quantifying PAI-1 levels in human breast tumor tissue. This assay combines pressure-cycling technology to extract PAI-1 from breast tumor tissue with a highly sensitive liposome polymerase chain reaction immunoassay for quantification of PAI-1 in the tissue extract. The new PAI-1 assay method reduced the total assay time to one day and improved assay sensitivity and dynamic range by >100, compared to ELISA.
Keywords: breast cancer, plasminogen activator inhibitor type-1, tissue biomarkers, high-pressure tissue extraction, immunoliposome-PCR, immunoassay
Introduction
The identification of patients at high risk for recurrence is one of the most important assessments in the clinical management of breast cancer. About 70% of early-stage node-negative breast cancer is cured by locoregional surgery and radiotherapy supplemented, when appropriate, with endocrine therapy [1, 2]. These patients receive no benefit from adjuvant chemotherapy. However, 30% of node-negative patients will experience recurrence and this rate is reduced significantly if these patients receive adjuvant chemotherapy [3]. Histomorphometric characteristics, such as tumor size, grade, and steroid receptor status are not effective in identifying patients at high risk of recurrence [4]. This creates a dilemma for clinicians when deciding on the appropriate choice of treatment, particularly for grade-2 (G2) tumors.
The urokinase plasminogen activator system [5-7] includes urokinase-type plasminogen activator (uPA), urokinase-type activator receptor (uPAR, CD87) and the uPA inhibitor, plasminogen activator inhibitor type-1 (PAI-1). These serine proteases modulate cell adherence to the extracellular matrix and are involved in both normal tissue repair and malignancy [8, 9]. In tumor cells uPA expression is up-regulated by a loss of transcriptional control [10]. Binding of uPA to its receptor leads to localized proteolysis of the extracellular matrix thereby promoting tumor cell proliferation, migration, and invasion [7, 8]. The aberrant expression of uPA by tumor cells has been implicated in other aspects of malignant transformation including angiogenesis, tumor cell extravasation and intravasation, and proliferation of tumor cells at metastatic sites [7, 11, 12].
In normal physiology, uPA proteolysis is tightly regulated, principally by PAI-1 [13]. Thus, it is paradoxical that elevated PAI-1 levels are correlated with reduced disease-free survival (DFS) and overall survival (OS) in breast cancer [14]. Recent studies have suggested several mechanisms whereby levels of PAI-1 in excess of those required for suppression of uPA activity may promote malignancy and portent a poor prognosis. Elevated levels of PAI-1 can promote angiogenesis [15] and stimulate tumor cell proliferation [6]. Elevated levels of PAI-1 also appear to inhibit apoptosis [16] leading to enhanced tumor cell survival. The anti-fibrionlytic action of PAI-1 appears to facilitate the attachment of circulating tumor cells to the endothelium, which can lead to extravasation and the formation of metastases [14, 17]. PAI-1 has a higher binding affinity for the B domain of vitronectin than uPAR [18]. Consequently, at high concentrations, PAI-1 can disrupt uPAR-vitronectin interaction resulting in tumor cell detachment from the extracellular matrix promoting tumor cell dissemination and metastasis [19]. PAI-1 may also promote detachment, migration, and reattachment of tumor cells to their substratum by sustaining mitogenic tumor cell stimuli initiated by the uPA/uPAR complex [20].
Jänicke et al. [21-23] were the first to demonstrate that determination of the uPA and PAI-1 levels in primary breast tumor tissue extracts by enzyme-linked immunosorbent assay (ELISA) yielded strong prognostic information regarding DFS and OS in patient with newly diagnosed node-negative breast cancer. These original studies established the independent statistically validated prognostic utility of uPA and PAI-1 and used a likelihood method, based upon DFS, to determine prognostic cut-off values of 3 ng/mg of protein for uPA and 14 ng/mg of protein for PAI-1 [24-26]. The original work by Jänicke has been validated by a number of clinical trials. The first large-scale study was the Chem-N0 node-negative prospective randomized multicenter therapy trial conducted from 1993 to 1998 with 647 registered patients from 12 academic centers in Germany and Slovenia [25, 27]. Retrospective analysis of the Chem-N0 trial found that the recurrence rate for the high uPA/PAI-1 observation group was 23% versus 12.9% for the low uPA/PAI-1 group. The high-risk uPA/PAI-1 patients receiving adjuvant chemotherapy had a 26% lower estimated probability of disease recurrence than those randomized for observation [3]. The prospective randomized NNBC-3 trial was conducted from 2002 to 2009 with 4,150 registered patients from 153 centers in Germany and France [28]. One important finding of this trial was the ability of uPA and PAI-1 to successfully classify G2 breast cancer patients into high- and low-risk groups. These trials, combined with a meta-analysis of 8,377 patients from published databases [29], have established uPA and PAI-1 as the only breast cancer biomarkers to reach the highest level of evidence (LOE-1) [30]. They are also considered to be the only biomarkers appropriate for the routine assessment of prognosis in patients with newly diagnosed node-negative breast cancer according to the American Society of Clinical Oncology (ASCO) guidelines [31]. The prognostic benefit of uPA and PAI-1 appears to be independent of tumor size, grade, axillary node status, and steroid receptor status [25, 32]. A recent study by Descotes et al. [33] concluded that poor prognosis related to uPA is only valid when high levels of PAI-1 are also present, and that the clinical relevance of PAI-1 is not improved by uPA.
The above clinical trials employed colorimetric ELISA of breast tumor tissue extracts to measure uPA and PAI-1 levels, which were then normalized to the protein content of the extracts [34]. Current ASCO guidelines recommend 100 to 300mg of fresh or frozen breast cancer tissue for determination of uPA and PAI-1 levels by ELISA [31, 35]. The need for this quantity of fresh or frozen breast tumor tissue requires a surgical biopsy or vacuum-assisted core biopsy with an 8-gauge needle [36] and precludes the use of 14-gauge needle-core biopsies that are more common in clinical practice [37]. It also imposes a lower limit on the size of analyzable tumors that is well above the detection limit of modern radiological methods Thomassen et al [38] used ELISA to compare normalized uPA and PAI-1 levels measured in 10-30mg core biopsy specimens and 90-300 mg tumor tissue taken from the same specimens immediately after surgery. ELISA measurements using the smaller tissue specimens correctly classified risk in 95% of the patients surveyed; however, correlation between individual uPA and PAI-1 levels in the small biopsy specimens versus the larger tissue samples was only 0.789 and 0.901, respectively. Both uPA and PAI-1 are involved in normal tissue-remodeling; consequently, pre-operative diagnostic core needle biopsies can alter their levels [39]. It has recently been demonstrated that core biopsies lead to a fibroblastic inflammatory response that results in increased uPA and PAI-1 levels that can persist for up to 9 days in the surrounding tissue and even longer in the area immediately adjacent to the biopsy channel. Consequently, tissue biopsies taken at the time of surgery for assessment of uPA and PAI-1 could prove unreliable if earlier pre-operative core needle biopsies had been performed [39].
Unfortunately, no other method to quantify uPA and PAI-1 levels in tissues has proven to be a reliable substitute for the ELISA assay. Although uPA and PAI-1 can be detected by immunohistochemistry (IHC) using frozen or formalin-fixed, paraffin-embedded (FFPE) tissues, a correlation between protein levels, IHC scoring, and patient outcome was not found [32, 40, 41]. In addition, protein concentrations cannot be reliably determined by IHC due to saturation of staining at relatively low protein levels [42]. Preliminary studies using reverse-phase protein arrays (RPPAs) for uPA and PAI-1 have shown promise, but are not yet established as being quantitatively accurate [41]. Quantification of mRNA has also proven to be an unreliable substitute for ELISA in assessing uPA and PAI-1 levels [13, 42-44]. A direct comparison of ELISA protein levels and reverse-transcriptase polymerase chain reaction (RT-PCR) quantification of uPA and PAI-1 mRNA in tumor tissues resulted in a concordance of only 84% for uPA and 70% for PAI-1 [42]. Grebenchtchikov et al [44] demonstrated that plasma levels of PAI-1 did not reflect its concentration in tumor tissue, thus serum PAI-1 levels are not recommended for assessing prognosis in breast cancer.
Finally, the commercially available FEMTELLE ELISA assays for uPA and PAI-1 [35] in clinical use require three days to perform and have limited dynamic range and sensitivity. In this study we describe a new assay method for tissue biomarkers using PAI-1 from human breast tissue as an example. This assay combines pressure-cycling assisted extraction of PAI-1 from breast tumor tissue with a highly sensitive immunoliposome polymerase chain reaction (ILPCR) assay [45] for quantification of PAI-1 in the tissue extract. Using this new assay method, the total assay time for PAI-1 is reduced to one day and the dynamic range and sensitivity is increased by >100, compared to ELISA.
Materials and Methods
Tissues
Frozen human breast tumor tissue was obtained from the Armed Forces Institute of Pathology Breast Tissue Registry under an Institutional Review Board approved protocol (IP1872C-C1). The study used existing tissue samples that were originally taken during normal medical procedures and not for research purposes. All tissue samples used in this study were stripped of patient identifiers (de-identified) prior to the current study. No code-key linking the de-identified tissues to their patient identifiers was maintained. This study is only concerned with PAI-1 assay development, thus no patient characteristics or outcomes were reported. Upon completion of the study, all remaining tissue was disposed of as hazardous waste.
Materials
The PAI-1 FEMTELLE ELISA kit was purchased from Sekisui Diagnostics, LLC (Stamford, CT, USA). Pancreatic DNase I, cholesterol, bovine serum albumin (BSA) fraction V (RIA grade), phosphate-buffered saline (PBS; 10 mM potassium phosphate, 2.7 mM KCl, 137 mM NaCl, pH 7.4) , Tris-buffered saline (TBS; 50 mM Tris, 150 mM NaCl, pH 7.6), Triton X-100 (ultra-grade), HEPES buffer, citrate buffer, DEAE-Sepharose CL-6 B, and polyethylene glycol bisphenol A epichlorohydrin copolymer (PEG copolymer) were obtained from Sigma-Aldrich (St. Louis, MO, USA). The phospholipids used to prepare the detection liposomes were obtained from Avanti Polar Lipids (Alabaster, AL, USA). Absolute ethanol was purchased from Pharmco-AAPER (Brookfield, IL, USA). Polycarbonate membranes (13mm diameter, 0.1-micron pore size) were purchased from Costar Corporation (Cambridge, MA, USA). The DNA reporter encapsulated within the liposomes was an 84-base segment derived from the human β2-microglobin transcript. This segment spans an intron and thus is unlikely to be present in human tissue. The reporter DNA was prepared by cloning β2-microglobin cDNA, prepared and amplified from HeLa cell RNA, into a pCR2.1-TOPO T/A plasmid vector, which was used to transform One-Shot E. Coli. All cloning reagents and kits were purchased from Life Technologies-Invitrogen (Grand Island, NY, USA). A detailed description of reporter preparation using this method has been published previously [46]. NeutrAvidin, casein, and the bicinchoninic acid (BCA) protein assay kit were purchased from Thermo Fisher-Pierce Biotechnology (Rockford, IL, USA). Primers were purchased from Integrated DNA Technologies (Coralville, IA, USA). TaqMan universal PCR Master Mix, AmpliTaq Gold, and the Taqman probes for quantitative real-time PCR (qPCR) were purchased from Life Technologies-Applied Biosystems (Grand Island, NY, USA). FT500 pulse tubes for pressure-based disruption of tissues were obtained from Pressure Biosciences, Inc. (South Easton, MA, USA). General reagents, solvents, and laboratory supplies were obtained from Sigma-Aldrich.
Instruments
Homogenization of frozen tissue was performed using a Mikro Dismembrator model S from Thermo-Fisher Scientific (Pittsburgh, PA, USA). Pressure-based tissue extraction was performed on a Barocycler NEP2320 from Pressure Biosciences, Inc (South Easton, MA). A Spectra Max M5 microplate reader (Molecular Devices, Sunnyvale, CA, USA) was used for optical absorbance measurements. qPCR was carried out on an ABI 7500 from Life Technologies-Applied Biosystems. Microwell plates were washed using a BioTek model ELx405 automated plate washer (Winooski, VT, USA). Liposome sizing by extrusion through polycarbonate membranes was carried out with a temperature-jacketed Thermobarrel Extruder from Lipex Biomembranes (Vancouver, Canada). Temperature control during extrusion was achieved by connecting a circulating water bath to the water jacket of the extruder.
Commercial PAI-1 ELISA Assay
Tissue extraction and ELISA for PAI-1 was carried out as described by Schmitt et al [35] and the instructions provided with the FEMTELLE kit (product number 821) from Sekisui Diagnostics. Briefly, frozen specimens equivalent to 100 to 300 mg of wet tissue were homogenized in 1.8 mL of TBS using the Sartorius Mikro Dismembrator. A volume of 0.2 mL of 10% (w/v) Triton X-100 in TBS was added to the tissue homogenate to yield a detergent concentration of 1% (w/v). The homogenate was then stirred for 16 hours at 4°C. The resulting suspension was centrifuged at 100,000 x g for 60 min at 4°C and the clear supernatant was decanted. The total protein concentration was measured using a BCA protein assay and was adjusted to 2-3.5 mg/mL using TBS. The samples were diluted 1:20 (v/v) in sample buffer [1% (w/v) BSA, 0.4% (w/v) Triton X-100] in PBS prior to the ELISA assay.
Following tissue extraction, the ELISA colorimetric assay was performed over two consecutive days. On the first day, 100 μL of PAI-1 standards, diluted specimens, and controls were added, in duplicate, to microwells coated with a murine anti-human PAI-1 capture antibody. The microwell strips were covered and incubated for 16-20 hrs at 4°C. On the second day, the microwells were washed 4-times with wash buffer [0.4% (w/v) Triton X-100 in PBS, pH 7.4]. A 100 μL aliquot of biotinylated monoclonal anti-human PAI-1 detection antibody was added to each microwell and the strips were sealed and incubated at room temperature for 1 hr. The microwells were then washed as described above. A 100 μL aliquot of enzyme conjugate was added to each microwell and the strips were covered and incubated at room temperature for 1 hr. The microwells were washed again as described above. The enzyme conjugate was Streptavidin-Horseradish peroxidase. Each microwell then received 100 μL of substrate solution (TMB; perborate/3,3',5,5'-tetramethylbenzidine) and the wells were covered and incubated for 20 min at room temperature. The reaction was stopped by adding 50 μL of 0.5 N sulfuric acid and the absorbance of the microwells was read on the plate reader at 450 nm within 10 min. A standard curve was prepared by plotting the absorbance of the PAI-1 standards versus their respective concentrations.
Preparation of Liposome Detection Reagent
Methods for the preparation, purification, and characterization of the liposome detection reagent have been published previously [45]. Briefly, liposomes were prepared by mixing chloroform solutions of 1,2-distearoyl-sn-glycero-3-phosphocholine (24.5 mol %), cholesterol (45 mol %), 1,2-dioleoyl-3-dimethylammoniumpropane (25 mol %), 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)2000] ammonium salt (4.75 mol %), 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[biotinyl(polyethylene glycol)2000] ammonium salt (0.25 mol%), and lissamine rhodamine B-1,2-dihexadecyl-sn-glycero-3-phosphoethanolamine triethylammonium salt (0.5 mol%). The solvent containing the lipid mixture was evaporated by drying under a stream of N2, and then under high vacuum for at least 4 h. The dried lipid film was hydrated in 300 mM citrate buffer, pH 4, by vortexing the suspension at 65°C. The resulting multilamellar vesicles were then subjected to five freeze/thaw cycles using liquid nitrogen and a water bath set to 65°C. The liposomes were extruded 10 times through two stacked 0.1-micron polycarbonate membranes at 65°C using the vesicle extruder, which led to the formation of unilamellar liposomes ~100 nm in diameter. Ethanol was slowly added to the rapidly vortexed liposome suspension until the final ethanol concentration was 40% by volume. The reporter DNA (300 μg/mg total lipid) was then added to the lipid suspension, which was incubated at 40°C for 1 h. The resulting liposomes were dialyzed against 2 L of the citrate buffer followed by 2 L of 20 mM HEPES, 145 mM NaCl, pH 7.5. Unencapsulated reporter DNA was removed by ion-exchange gel filtration on DEAE-Sepharose CL-6 B (0.5 ml of gel/mg total lipid) using HEPES buffer.
Pressure-Assisted Tissue Extraction
Frozen tissue specimens equivalent to 1, 10, 50, or 100 mg of wet tissue were homogenized in 1.8 mL of TBS using the Sartorius Mikro Dismembrator. A volume of 0.2 mL of 10% (w/v) Triton X-100 in TBS was added to the tissue homogenate to yield a detergent concentration of 1% (w/v). The homogenate was then added to a FT500 pulse tube and placed in the Barocycler instrument. A pressure-cycling program was run that consisted of 40 pressurization cycles carried out at 4°C. Each pressurization cycle consisted of 20 sec at 35,000 psi followed by 10 sec at atmospheric pressure. The transitions between 35,000 psi and ambient pressure occurred over ~2 sec. The total processing time was about 25 min. The pulse tube was then removed from the instrument and vortexed for 30 sec. The suspension was centrifuged at 100,000 x g for 60 min at 4°C, and the clear supernatant was decanted. The total protein concentration was measured using a BCA protein assay and the concentration was adjusted to 2-3.5 mg/mL using TBS buffer. For the ILPCR assay the samples were diluted 1:20 (v/v) in sample buffer as described above.
ILPCR Assay
The ILPCR assay was performed using the commercial FEMTELLE ELISA kit as describe above up to the step where the Streptavidin-horseradish peroxidase was added. The single exception was that the antigen or tissue sample was incubated in the microwells for 2 hr at 37°C rather than overnight at 4°C. In place of the Streptavidin-conjugate a volume of 100 µL of NeutrAvidin (2 μg/mL) in PBS was added to each microwell and the plate was incubated at 37°C for 1 h. The solution was aspirated and the wells were washed twice with 300 µL of PBS. The plate wells were then blocked with 1% (w/v) casein in PBS and washed as described above. A volume of 100 µL of liposome detection reagent at a concentration of 100 nM (0.1 nmol total lipid/ml) in 1% (w/v) PEG copolymer in PBS was added to each well and the plate was incubated at room temperature for 1 h. The microwells were then washed as describd above. Each well then received 100 µl of DNase I (10 U/well) in 10 mM CaCl2, 10 mM MgCl2, 20 mM HEPES, pH 7.8, to degrade any unencapsulated DNA. The digestion was carried out at 37°C for 20 min, and the DNase I was then inactivated by heating the plate at 80°C for 10 min. The wells were washed 5 times with 300 µl of PBS. Finally, the liposome detection reagent was lysed by the addition of 100 µl of 10 mM Triton X-100 in 10 mM borate, pH 9.0, followed by incubation at room temperature for 20 min on a plate shaker at 600 rpm. Following lysis of the liposomes, a 1-μL aliquot from each microwell was added to 12.5 μl of 2x TaqMan Universal PCR Master Mix. Each PCR tube then received 1 µL of forward and reverse primers (15 µM each), and 1 µL of the probe (5 µM). Water was then added to bring the reaction volume to 25 μL. qPCR was preceded by a 2-min UNG incubation step at 50°C and a 10-min AmpliTaq Gold activation step at 95°C. Forty cycles of PCR were then performed, where each cycle consisted of a 15-sec denaturation step at 95°C and a 1-min annealing/extension step at 60°C [45]. The sequences of the primers, the probe, and the DNA reporter have been published previously [45, 46]. A standard curve was prepared by plotting the cycle threshold values (Ct) versus the log of their respective concentrations.
Results
Extraction of PAI-1 from Breast Tumor Tissue
We first performed a study to determine if pressure-cycling extraction would improve the recovery of PAI-1 from frozen breast tumor tissue. A frozen breast tumor specimen, equivalent to 400 mg of wet tissue, was homogenized for 30 sec in 1.8 mL of TBS using the Sartorius Mikro Dismembrator. A volume of 0.2 mL of 10% (w/v) Triton X-100 in TBS was added to the tissue homogenate to yield a detergent concentration of 1% (w/v). The homogenate was then divided into four equal aliquots. Two aliquots were processed using the standard Sekisui Diagnostics method [35], which included overnight stirring for 16 hours at 4°C.
The remaining two aliquots were processed in the Barocycler instrument using a pressure-cycling program consisting of 40 pressurization cycles carried out at 4°C. Each pressurization cycle consisted of 20 sec at 35,000 psi followed by 10 sec at atmospheric pressure. The total protein concentration of the four aliquots was adjusted to 2.5 mg/mL with TBS. The PAI-1 concentration of each specimen was then determined using the ELISA assay format and the results from the two samples for each extraction method were averaged. The results are shown in Table 1. A t-test of the data showed that the total protein and PAI-1 recovered was statistically identical for the two methods. However, the total processing time required was reduced by about 16 hr (87%) by using the pressure-cycling tissue protein extraction method.
Table 1
Recovery of protein from frozen human breast tumor tissue
| Parameter | Standard Methoda | Pressure-Cycling Methodb |
|---|---|---|
| Total Protein Recoveredc | 2.8 ± 0.22 mg/mL | 3.0 ± 0.27 mg/mL |
| PAI-1 Recovered | 68 ± 7.4 ng/mL | 75 ± 6.8 ng/mL |
| Normalized PAI-1 Recovery | 24 ± 3.3 ng/mg | 25 ± 3.2 ng/mg |
| Total Processing Time | 18 hr | 2.3 hr |
aThe standard method used overnight stirring of the homogenate as described in the text. bThe pressure-assisted method consists of high-pressure cycling on a commercial Barocycler NEP2320 using FT500 pulse tubes as described in the text. cTotal protein was measured using the BCA colorimetric protein assay.
ILPCR Assay for PAI-1 in Breast Tumor Tissue Extracts
Next, we performed a series of studies to determine the ability of an ILPCR assay for PAI-1 to accurately quantify PAI-1 levels in soluble breast tumor tissue extracts. A diagram of the liposome detection reagent used in the ILPCR assay is shown in Figure 1. In our initial experiment the PAI-1 concentration standards from the Sekisui Diagnostics FEMTELLE PAI-1 ELISA assay kit were each reconstituted in 1.0 mL of deionized water to produce PAI-1 assay standards of 1.0, 2.5, 5.0, 7.5, and 10 ng/mL. These standards were analyzed using the ELISA kit by following the manufacturer's instructions as outlined in the Methods. The assay standards and a water blank (100 μL each) were added to the wells, in duplicate, and incubated overnight at 4°C. The next day biotinylated detection antibody was added followed by streptavidin-horseradish peroxidase conjugate and TMB developing solution. After 20 min, sulfuric acid solution was added to stop the development reaction and the absorbance was measured at 450 nm. The resulting dose-response curve, which closely matched the one provided by the manufacturer, is shown in Figure 2A. Regression analysis yielded a correlation coefficient of 0.997 using a 2nd-order polynomial fit. The limit of detection (LOD), reported by the manufacturer, was 125 pg/mL and the dynamic range was ~100.
Figure 1
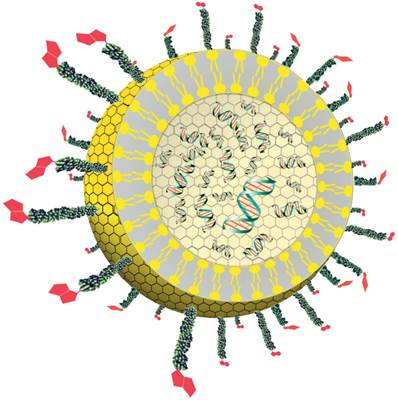
Diagram showing a cross-section of the liposome detection reagent. The reporter DNA (green and red) is encapsulated inside the liposome (yellow). The polyethylene glycol polymers (green) with biotin receptors (red) are conjugated to phospholipids located within the outer surface of the liposome.
An ILPCR assay using the above PAI-1 standards was then performed with the following modifications. First, additional standards of 100, 10, and 1 pg/mL were prepared by serial dilution of the 1 ng/mL PAI-1 standard. All of the standards and a water blank (100 μL) were then added to the microwells of the Sekisui Diagnostics plate strip, in duplicate, and incubated for 2 hr at 37°C. The biotinylated second antibody was added followed by NeutraAvidin and then the detection liposomes as described in the Methods section. Each well was treated with DNase I for 15 min and the enzyme was then heat deactivated at 80°C for 10 min. The detection liposomes were ruptured with 1% Triton X-100 and the released DNA reporter was quantified by qPCR. The resulting Ct values were plotted against the log of the PAI-1 molar concentration to produce the dose-response curve shown in Figure 2B. The plot was linear over the entire concentration range yielding a dynamic range of 10,000, which was >100-times greater than that of the ELISA assay. The apparent LOD for the assay (the lowest concentration measured) was 1 pg/mL, which was >100-times lower than that of the ELISA assay. The red line in Figure 1B is the assay blank and the dotted magenta line is the detection threshold defined as the Ct value of the blank minus 3-times the standard deviation of the blank (vertical blue bars). The Ct value for the PAI-1 concentration of 1 pg/mL was ~25, which is 2 Ct values below the detection threshold (Ct ~27) of the assay. It is important to emphasize that the ILPCR assay results were achieved by incubation of the standards at 37°C for 2 hr, whereas the ELISA assay required an overnight incubation at 4°C. This reduced assay time from two days to one day. Finally, little attempt was made to optimize the ILPCR assay as both the microplate substrate and the blocking reagent used in the FEMTELLE assay are not ideal for ILPCR assays. Thus, we anticipate that an ILPCR assay optimized for PAI-1 will show enhanced performance.
In a second experiment, 300 mg of fresh frozen tumor tissue was pulverized in 1.8 mL of TBS using the Sartorius Mikro Dismembrator. A volume of 0.2 mL of 10% (w/v) Triton X-100 in TBS was added to the tissue homogenate to yield a detergent concentration of 1% (w/v). The tissue was allowed to thaw at 4°C and then converted in a homogeneous suspension using a dounce homogenizer. The above process was repeated and the two homogeneous tissue suspensions were combined. From this suspension, aliquots of 150, 100 (two samples), 50, 10, and 1 mg of tumor tissue were prepared, each in 2 mL of 1 % (w/v) Triton X-100 in TBS. The 150 mg aliquot and one 100 mg aliquot were further processed and assayed for PAI-1 content using the standard Sekisui Diagnostics ELISA kit. The remaining tissue aliquots were subjected to pressure-assisted protein extraction and the PAI-1 content was assayed by ILPCR. The total protein content of all tissue aliquots was measured using the BCA protein assay kit.
A plot of PAI-1 content (ng/mL) versus tumor tissue mass is shown in Figure 3A. The PAI-1 concentrations shown were not corrected for dilution of the tissue extract. Results for both the standard ELISA assay (150 and 100 mg of tissue) and the ILPCR assay with pressure-assisted protein extraction (100, 50, 10, and 1 mg of tissue) are plotted on the same graph. The total protein concentration in the extract from the 150 mg tissue sample was 3.41 mg/mL and the corresponding PAI-1 concentration was 2.82 ng/mL. The most dilute tissue sample analyzed (1 mg) yielded a total protein extract with a concentration of 35 ng/mL and a corresponding PAI-1 concentration of 34 pg/mL. Tumor tissue samples of 100 mg were analyzed by both the ELISA and ILPCR assays, the latter using pressure-assisted protein extraction. The two methods yielded similar results with total protein extract concentrations of 2.16 and 2.19 mg/mL and PAI-1 concentrations of 2.31 and 2.23 ng/mL for the ELISA and ILPCR methods, respectively. The data was fit by liner regression, which yielded a correlation coefficient of 0.996. This demonstrates that the two assay methods are effectively equivalent and that the ILPCR assay method does not deviate from linearity for tumor tissue masses down to 1 mg.
The PAI-1 concentrations were corrected for dilution and normalized to the total protein concentration of the tissue extracts and plotted in Figure 3B. The horizontal line in the plot is the average of the normalized PAI-1 concentrations over the range of 1 to 150 mg of tumor tissue, which was18.6 ng/mg ± 1.47 (standard deviation). All of the normalized PAI-1 concentrations were within ±10% of the mean. The data was also fit with a linear regression (not shown), which yielded a slope of -0.012 ng PAI-1 per mg of tumor tissue.
Figure 2
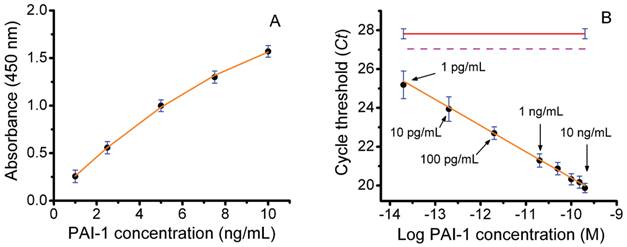
Assay of PAI-1 concentration standards by colorimetric ELISA and ILPCR. A) Colorimetric ELISA using the Sekisui Diagnostics ELISA assay kit performed by following the manufacturer's instructions. B) ILPCR assay performed suing PAI-1 standards from the ELISA assay, but with additional serial dilutions of 100, 10, and 1 pg/mL. The Streptavidin-HRP conjugate with colorimetric development was replaced with NeutrAvidin plus detection liposomes with qPCR quantification. Data points (black dots), standard deviations (blue bars), regression analysis (solid orange plot lines), ILPCR assay blank (solid red line), IPCR assay threshold (magenta dotted line).
Figure 3
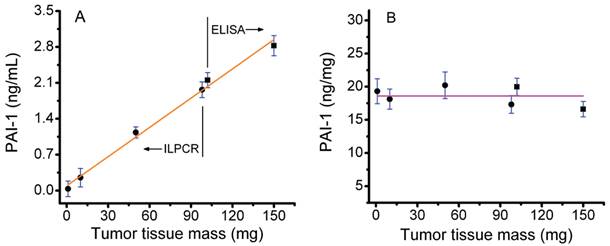
Assay of PAI-1 in human breast tumor tissue extracts. A) Conventional ELISA and ILPCR assays of PAI-1 concentration in total protein extracts derived from 1 to 150 mg of tumor tissue. Total protein extracts were prepared by conventional overnight stirring at 4°C for the ELISA assay and by pressure-assisted protein extraction for the ILPCR assay. B) The PAI-1 values plotted after being normalized to the total protein concentration of the corresponding tissue extracts. Results from the ELISA assay as shown as black squares and results from the ILPCR assay are shown as black dots. Standard deviations are shown as horizontal blue bars. The orange line in A shows the linear regression fit of the data and the magenta line in B is the average normalized PAI-1 value for all of the tumor tissue samples analyzed.
Discussion
In this study we report an improved assay method for quantifying PAI-1 in frozen human breast tumor tissue specimens. The determination of tissue biomarkers, such as PAI-1, is a two-step process consisting of homogenization and extraction of the biomarker from fresh or frozen tissue followed by quantification of the biomarker in the tissue extract by immunoassay. The method described in the current study includes improvements in both the extraction and immunoassay steps. The result is a reduction in assay time from 3 days to 1 day, a >100-fold improvement in assay sensitivity, and a >100-fold improvement in assay dynamic range.
The improvement in assay time was achieved by using a pressure-cycling method that accelerates cell lysis by rapidly cycling between ambient pressure and 35,000 psi. Tissue protein homogenization and extraction was achieved in a total time of 2.3 hr instead of the 20 hours required for the conventional FEMTELLE assay procedure. The temporal improvement in the detergent extraction step alone was even more dramatic; pressure-cycling reduced this time from 18 hr to 25 min. Although pressure-cycling reduced the time required for tissue extraction the yield of total protein and PAI-1 was statistically identical to that achieved using the conventional overnight extraction method. This reinforces previous reports that pressure-cycling promotes rapid disruption of cell membranes [49-52]. The greater efficiency of the pressure-based method is likely due to both the physical shear stress and gas cavitation produced by rapidly cycling between 35,000 psi and ambient pressure [49, 48]. Pressure-cycling technology has been shown to enhance the recovery of proteins from complex tissue matrices including formalin-fixed human aortas [49], formalin-fixed, paraffin-embedded archival tissues [50], and membrane proteins from ovarian cancer tissue [51]. The significant improvement in the time required to extract of PAI-1 from frozen human breast tumor tissue observed in the current study is consistent with these previous reports [52]. We found that cryogenic homogenization of frozen breast tissue was necessary for successful protein extraction by pressure-cycling. Extraction efficiency was significantly reduced when diced frozen tissue was used in place of tissue homogenized with the dismembrator. A similar finding was reported in the application of pressure-cycling to extract membrane proteins from ovarian cancer tissue [51].
A second improvement in the PAI-1 assay method was the replacement of ELISA with ILPCR. Specifically, the Streptavidin-HRP conjugate detection reagent was replaced by liposomes with encapsulated reporter DNA and biotin-labeled PEG polymers on their outer surface. The biotin-labeled liposomes were coupled to the biotin-labeled secondary antibodies from the FEMTELLE kit using Neutravidin. Consequently, qPCR replaced the optical density of the HRP substrate (TMB) as the detection method. This resulted in a PAI-1 immunoassay with significantly improved sensitivity and dynamic range [45]. An additional advantage of the increased sensitivity of ILPCR was the elimination of the overnight antigen incubation step required in the ELISA method. Satisfactory sensitivity with ILPCR was achieved using incubation at 37 C for 1 hr, thus reducing the total assay time by one day. The apparent limit of detection of the ILPCR assay was 1 pg/mL of PAI-1. Figure 1 shows that the Ct value for this concentration was well below that of the assay detection threshold. This suggests that the true detection limit of the ILPCR assay is likely below 1 pg/mL of PAI-1, the lowest concentration that was measured in this study. Additional assay controls to guard against false-positive results due to contaminating DNA or false negative results due to PCR inhibitors or pre-mature rupture of the liposomes can be included in the ILPCR assay as discussed in Mason et al. [45]. Although we did not develop an assay for uPA in this study, the combination of pressure-cycling for tissue extraction and the ILPCR assay method for quantification are anticipated to result in a level of improvement similar to that obtained for PAI-1.
A study by Thomssen et al. [38] showed that normalized PAI-1 values correctly classified 95% of patients for risk of recurrence using 10-30 mg of breast biopsy tissue. The current study further reduces the required amount of breast cancer tissue to below 1 mg. This would allow the determination of PAI-1 levels in individual 14- to 16-guage needle cores obtained without vacuum assist [36] and in frozen thin tissue sections. The increased sensitivity of the ILPCR assay would also allow PAI-1 levels to be measured in very small tumors, below the current limits of radiologic detection, but detectable by histology [53, 54]. When evaluating larger tumors, measurements on small needle cores can mask tumor heterogeneity; consequently, evaluation of multiple biopsy specimens would still be necessary.
There is a clear disadvantage to the requirement for fresh or frozen breast tumor tissue to determine PAI-1 levels using the ELISA method. Although IHC is has proven insufficiently accurate to determine PAI-1 levels in FFPE tissues, preliminary studies using the RPPA method have shown promise [41]. Like ELISA, the RPPA method uses antibodies coupled with colorimetric detection. The sensitivity and dynamic range of RPPA assays for PAI-1 could potentially be increased by substituting ILPCR for protein detection. Pre-coupling of antibodies to the liposomes [45] would allow for multiplexing by associating each antibody with a unique DNA reporter sequence. This would allow the quantification step to be decoupled from the protein array resulting in improved sensitivity and dynamic range for each analyte.
In summary, we have presented an improved assay method for the quantification of the biomarker PAI-1 in human breast tumor tissue that combined pressure-cycling tissue extraction with the ILPCR assay format. The result was a reduction in assay time from three days to one day and improvements in assay sensitivity and dynamic range of >100. This improved assay method should be applicable to uPA and other tissue protein biomarkers.
Acknowledgements
This project was supported, in part, by funds from the Veterans Health Administration under Merit Review Award VA 1101BX001092-01 (J.T.M.). The content of this publication does not necessarily reflect the views of the Veterans Health Administration, nor does mention of trade names, commercial products, or organizations imply endorsements by the United States Government. The authors thank Junkun He, PhD, for performing many of the immunoassays described in this study.
Competing Interests
Carol B. Fowler and Jeffrey T. Mason are named as lead contributors on two patents (USPTO numbers: 20130011854 and 20100136613) entitled “Pressure-Assisted Antigen Retrieval (PAAR), Pressure-Assisted Molecule Recovery (PAMR), and Pressure-Assisted Tissue Histology (PATH)”. Jeffrey T. Mason is named as a lead contributor on two patents (USPTO numbers: 20090176250 and 20050158372) entitled “Immunoliposome-Nucleic Acid Amplification (ILNAA) Assay”. The assignee on all four patents is the United States of America as represented by the Secretary of the Army and/or the Secretary of Veterans Affairs.
References
1. Ulisse S, Baldini E, Sorrenti S. et al. The urokinase plasminogen activator system: a target for anti-cancer therapy. Curr Cancer Drug Targets. 2009;9:32-71
2. Harbeck N, Thomssen C. A new look at node-negative breast cancer. The Oncologist. 2010;15:29-38
3. Harbeck N, Schmitt M, Meisner C. et al. Ten-year analysis of the prospective multicenter Chem-N0 trial validates American Society of Clinical Oncology (ASCO)-recommended biomarkers uPA and PAI-1 for therapy decision making in node-negative breast cancer patients. Eur J Cancer. 2013;49:1825-1835
4. Jänicke F, Prechtl A, Thomssen C. et al. Randomized adjuvant chemotherapy trial in high-risk lymph node-negative breast cancer patients identified by urokinase-type plasminogen activator and plasminogen activator inhibitor type 1. J Natl Cancer Inst. 2001;93:913-920
5. Blasi F. Proteolysis, cell adhesion, chemotaxis, and invasiveness are regulated by the u-PA-u-PAR-PAI-1 system. Thromb Haemost. 1999;82:298-304
6. Duffy MJ. Urokinase plasminogen activator and its inhibitor, PAI-1, as prognostic markers in breast cancer: from pilot to level 1 evidence studies. Clin Chem. 2002;48:1194-1197
7. Andreasen PA, Kjøller L, Christensen L. et al. The urokinase plasminogen activator system in cancer metastasis: a review. Int J Cancer. 1997;72:1-22
8. Malinowsky K, Wolff C, Berg D. et al. uPA and PAI-1-related signaling pathways differ between primary breast cancer and lymph node metastases. Transl Oncol. 2012;5:98-104
9. Reuning U, Magdolen V, Wilhelm O. et al. Multifunctional potential of the plasminogen activation system in tumor invasion and metastasis. Int J Oncology. 1998;13:893-906
10. Nagamine Y, Medcalf RL, Muñoz-Cánoves P. Transcriptional and posttranscriptional regulation of the plasminogen activator system. Thromb Haemost. 2005;93:661-675
11. Choong PF, Nadesapillai AP. Urokinase plasminogen activator system: a multifunctional role in tumor progression and metastasis. Clin Orthop Relat Res. 2003;415:S46-58
12. Stephens RW, Brunner N, Jänicke F. et al. The uorkinase plasminogen activator system as a target for prognostic studies in breast cancer. Breast Cancer Res Treat. 1998;52:99-111
13. Witzel ID, Milde-Langosch K, Wirtz RM. et al. Comparison of microarray-based RNA expression with ELISA-based protein determination of HER2, uPA and PAI-1 in tumor tissue patients with breast cancer and relation to outcome. J Cancer Res Clin Oncol. 2010;136:1709-1718
14. Kwaan HC, Mazar AP, McMahon BJ. The apparent uPA/PAI-1 paradox in cancer: more than meets the eye. Semin Thromb Hemost. 2013;39:382-391
15. Hildenbrand R, Schaaf A, Dorn-Beineke A. et al. Tumor stroma is the predominant uPA-, uPAR, PAI-1-expressing tissue in human breast cancer: prognostic impact. Histol Histopathol. 2009;24:869-877
16. Balsara RD, Ploplis VA. Plasminogen activator inhibitor-1: the double-edge sword in apoptosis. Thromb Haemost. 2008;100:1029-1036
17. Prager GW, Breuss JM, Steurer S. et al. Vascular endothelial growth factor (VEGF) induces rapid prourokinase (pro-uPA) activation on the surface of endothelial cells. Blood. 2004;103:955-962
18. Okumura Y, Kamikubo Y, Curriden SA. et al. Kinetic analysis of the interaction between vitronectin and the urokinase receptor. J Biol Chem. 2002;277:9395-9404
19. Czekay RP, Aertgeerts K, Curriden SA. et al. Plasminogen activator inhibitor-1 detaches cells from extracellular matrices by inactivating integrins. J Cell Biol. 2003;160:781-791
20. Czekay RP, Loskutoff DJ. Unexpected role of plasminogen activator inhibitor 1 in cell adhesion and detachment. Exp Biol Med (Maywood). 2004;229:1090-1096
21. Jänicke F, Schmitt M, Ulm K. et al. Urokinase-type plasminogen activator antigen and early relapse in breast cancer. Lancet. 1989;2:1049
22. Jänicke F, Schmitt M, Hafter R. et al. The urokinase-type plasminogen activator (u-PA) is a potent predictor of early relapse in breast cancer. Fibrinolysis. 1990;4:69-78
23. Jänicke F, Schmitt M, Graeff H. Clinical relevance of the urokinase-type and tissue-type plasminogen activators and their type 1 inhibitor in breast cancer. Sem Thromb Hemost. 1991;17:303-312
24. Jänicke F, Pache L, Schmitt M. et al. Both the cytosols and detergent extracts of breast cancer tissues are suited to evaluate the prognostic impact of the uorkinase-type plasminogen activator and its inhibitor, plasminogen activator inhibitor type 1. Cancer Res. 1994;54:2527-2530
25. Jänicke F, Prechtl A, Thomssen C. et al. Randomized adjuvant chemotherapy trial in high-risk, lymph-node-negative breast cancer patients identified by urokinase-type plasminogen activator and plasminogen activator inhibitor type 1. J Natl Cancer Inst. 2001;93:913-920
26. Grøndahl-Hansen J, Christensen IJ, Rosenquist C. et al. High levels of urokinase-type plasminogen activator and its inhibitor PAI-1 in cytosolic extracts of breast carcinomas are associated with poor prognosis. Cancer Res. 1993;53:2513-2521
27. Harbeck N, Meisner C, Prechtl A. et al. Level-1 evidence for prognosis and predicative impact of uPA and PAI-1 in node-negative breast cancer provided by second scheduled analysis of multicenter Chem-N0 therapy trial. Breast Cancer Res Treat. 2001;69:213
28. Kantelhardt EJ, Vetter M, Schmitt M. et al. Prospective evaluation of prognostic factors uPA/PAI-1 in node-negative breast cancer: phase III NNBC3-Europe trial (AGO, GBG, EORTC-PBG) comparing 6xFEC versus 3xFEC/3xDocetaxel. BMC Cancer. 2011;11:140
29. Look MP, van Putten WL, Duffy MJ. et al. Pooled analysis of prognostic impact of urokinase-type plasminogen activator and its inhibitor PAI-1 in 8377 breast cancer patients. J Natl Cancer Inst. 2002;94:116-128
30. Schmitt M, Harbeck N, Brünner N. et al. Cancer therapy trials employing level-of-evidence-1 disease forecast cancer biomarkers uPA and its inhibitor PAI-1. Expert Rev Mol Diagn. 2011;11:617-634
31. Harris L, Fritsche H, Mennel R. et al. American Society of Clinical Oncology 2007 update of recommendations for the use of tumor markers in breast cancer. J Clin Oncol. 2007;25:5287-5312
32. Schmitt M, Harbeck N, Thomssen C. et al. Clinical impact of the plasminogen activator system in tumor invasion and metastasis: prognostic relevance and target for therapy. Thromb Haemost. 1997;78:285-296
33. Descotes F, Riche B, Saez S. et al. Plasminogen activator inhibitor type 1 is the most significant of the usual tissue prognostic factors in node-negative breast ductal adenocarcinoma independent of urokinase-type plasminogen activator. Clin Breast Cancer. 2008;8:168-177
34. Schmitt M, Mengele K, Napieralski R. et al. Clinical utility of level-of-evidence-1 disease forecast cancer biomarkers uPA and its inhibitor PAI-1. Expert Rev Mol Diagn. 2010;10:1051-1067
35. Schmitt M, Sturmheit AS, Welk A, Et al. Procedures for the quantitative protein determination of urokinase and its inhibitor, PAI-1, in human breast cancer tissue extracts by ELISA. Methods Mol Med. 2006;120:245-265
36. Wilson R, Kavia S. Comparison of large-core vacuum-assisted breast biopsy and excision systems. Recent Results Cancer Res. 2009;173:23-41
37. Schueller G, Jaromi S, Ponhold L. et al. US-guided 14-gauge core-needle breast biopsy: results of a validation study in 1352 cases. Radiology. 2008;248:406-413
38. Thomssen C, Harbeck N, Dittmer J. et al. Feasibility of measuring the prognostic factors uPA and PAI-1 in core needle biopsy breast cancer specimens. J Natl Cancer Inst. 2009;101:1028-1029
39. Haas S, Park TW, Hahne JC. et al. Influence of preoperative core biopsies on uPA/PAI-1 expression in breast cancer tissue. Virchows Arch. 2008;452:277-283
40. Ferrier CM, de Witte HH, Straatman H. et al. Comparison of immunohistochemistry with immunoassay (ELISA) for the detection of components of the plasminogen activation system in human tumor tissue. Br J Cancer. 1999;79:1534-1541
41. Malinowsky K, Böllner C, Hipp S. et al. UPA and PAI-1 analysis from fixed tissues - new perspectives for a know set of predicative markers. Curr Med Chem. 2010;17:4370-4377
42. Lamy PJ, Verjat T, Servanton AC. et al. Urokinase-type plasminogen activator and plasminogen activator inhibitor type-1 mRNA assessment in breast cancer by means of NASBA: correlation with protein expression. Am J Clin Pathol. 2007;128:404-413
43. Biermann J, Holzscheiter L, Kotzsch M. et al. Quantitative TR-PCR assays for the determination of urokinase-type plasminogen activator and plasminogen activator inhibitor type 1 mRNA in primary tumor tissue of breast cancer patients: comparison to antigen quantification by ELISA. Int J Mol Med. 2008;21:251-259
44. Grebenchtchikov N, Maguire TM, Riisbro R. et al. Measurements of plasminogen activator system components in plasma and tumor tissue extracts obtained from patients with breast cancer: an EORTC receptor and biomarker group collaboration. Oncol Rep. 2005;14:235-239
45. He J, Evers DL, O'Leary TJ. et al. Immunoliposome-PCR: a generic ultrasensitive quantitative antigen detection system. J Nanobiotechnology. J Nanobiotechnology. 2012;10:26
46. Mason JT, Xu L, Sheng ZM. et al. Liposome polymerase chain reaction assay for the sub-attomolar detection of cholera toxin and botulinum neurotoxin type A. Nature Protocols. 2006;4:2003-2011
47. Gross V, Carlson G, Kwan AT. et al. Tissue fractionation by hydrostatic pressure cycling technology: the unified sample preparation technique for systems biology studies. J Biomol Tech. 2008;19:189-199
48. Powell BS, Lazarev AV, Carlson G. et al. Pressure cycling technology in systems biology. Methods Mol Biol. 2012;881:27-62
49. Fu Z, Yan K, Rosenberg A. et al. Improved protein extraction and protein identification from archival formalin-fixed paraffin-embedded human aortas. Proteomics Clin Appl. 2013;7:217-224
50. Fowler CB, Waybright TJ, Veenstra TD. et al. Pressure-assisted protein extraction: a novel method for recovering proteins from archival tissues for proteomic analysis. J Proteome Res. 2012;11:2602-2608
51. Schneider LV, Likhte V, Wright WH. et al. Method for recovery and immunoaffinity enrichment of membrane proteins illustrated with metastatic ovarian cancer tissues. Int J Proteomics. 2012;2012:838630
52. Freeman E, Ivanov AR. Proteomics under pressure: development of essential sample preparation techniques in proteomics using ultrahigh hydrostatic pressure. J Proteome Res. 2011;10:5536-5546
53. Gruber IV, Rueckert M. et al. Measurement or tumor size with mammography, sonography and magnetic resonance imaging as compared to histological tumor size in primary breast cancer. BMC Cancer. 2013;13:328
54. Wilmes LJ, McLaughlin RL. et al. High-resolution diffusion-weighted imaging for monitoring breast cancer treatment response. Acad Radiol. 2013;20:581-591
Author contact
![]() Corresponding author: Jeffrey T. Mason, PhD, Laboratory of Proteomics and Protein Science, Baltimore Veterans Affairs Medical Center, Room 3C-139A, 10 North Greene Street, Baltimore, MD 21201, USA. Phone: 301-318-5626; Email: jeffrey.mason2gov
Corresponding author: Jeffrey T. Mason, PhD, Laboratory of Proteomics and Protein Science, Baltimore Veterans Affairs Medical Center, Room 3C-139A, 10 North Greene Street, Baltimore, MD 21201, USA. Phone: 301-318-5626; Email: jeffrey.mason2gov
Citation styles
APA
Fowler, C.B., Man, Y.G., Mason, J.T. (2014). An Ultra-Sensitive Immunoassay for Quantifying Biomarkers in Breast Tumor Tissue. Journal of Cancer, 5(2), 115-124. https://doi.org/10.7150/jca.8084.
ACS
Fowler, C.B.; Man, Y.G.; Mason, J.T. An Ultra-Sensitive Immunoassay for Quantifying Biomarkers in Breast Tumor Tissue. J. Cancer 2014, 5 (2), 115-124. DOI: 10.7150/jca.8084.
NLM
Fowler CB, Man YG, Mason JT. An Ultra-Sensitive Immunoassay for Quantifying Biomarkers in Breast Tumor Tissue. J Cancer 2014; 5(2):115-124. doi:10.7150/jca.8084. https://www.jcancer.org/v05p0115.htm
CSE
Fowler CB, Man YG, Mason JT. 2014. An Ultra-Sensitive Immunoassay for Quantifying Biomarkers in Breast Tumor Tissue. J Cancer. 5(2):115-124.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) License. See http://ivyspring.com/terms for full terms and conditions.