Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2013; 4(9):727-735. doi:10.7150/jca.7576 This issue Cite
Research Paper
SRPK1 Dissimilarly Impacts on the Growth, Metastasis, Chemosensitivity and Angiogenesis of Glioma in Normoxic and Hypoxic Conditions
Qianqian Wu*, Yingwei Chang*, Luping Zhang, Yuqiang Zhang, Ting Tian, Guoying Feng, Shuai Zhou, Qinyin Zheng, Fengchan Han ![]() , Fei Huang
, Fei Huang ![]()
Institute of Human Anatomy and Histology and Embryology, Otology & Neuroscience Center, Binzhou Medical University, 346 Guanhai Road, Laishan, Shandong Province, 264003, China.
* These authors contributed to the work equally and should be regarded as co-first authors.
Received 2013-9-3; Accepted 2013-10-19; Published 2013-11-21
Abstract
Glioma is among the ten most common causes of cancer-related death and has no effective treatment for it, so we are trying to find a new target for anticancer treatment. This study investigates the different expression of SRPK1 as a novel protein in glioma, which can influence tumor cells biological characteristics in normoxic and hypoxic environment. The expression levels of SRPK1 protein in glioma cell lines transfected with siSRPK1 or not were examined using immunofluorescence, RT-PCR and Western blot analysis, respectively. The impact of SRPK1 on the biological characteristics of U251 cells was further studied using methylthiazol tetrazolium assays, flow cytometry, and Transwell invasion chamber assays. The results showed that knockdown of SRPK1 inhibited tumor cells growth, invasion and migration in normoxic condition, but portion of the effect could be reversed in hypoxia. SRPK1 expression was induced in glioma cells by DDP treated, but not TMZ, in both normoxia and hypoxia conditions. We propose SRPK1 as a new molecular player contributing to the early treatment of glioma.
Keywords: SRPK1, glioma, growth, metastasis, chemosensitivity.
Introduction
Gliomas are the most common primary intracranial tumor [1]. The incidence of primary brain tumors is estimated to be 6 per 100,000 persons per year in China; despite aggressive treatment efforts patients are dead at a median of 14 months after diagnosis [2]. According to the World Health Organization classification, gliomas are graded from I to IV based on their degree of malignancy [3], about half of malignant gliomas in adults are glioblastomas multiforme (GBM) [4]. Unfortunately, a combination of radiation and chemotherapy to treat GBM is used and the median survival for GBM patients is still about 15 months [5]. Moreover, many of the patients have chemo- and radiotherapy resistance, causing very poor prognosis [6]. At this stage, we are not sure about of the glioma pathogenesis [3]; so many researchers have studied on the growth mechanism of malignant gliomas and try to find new effective methods for treatment.
Serine/arginine(SR/RS) protein kinases[7] family(SRPK) represent a class of enzymes that can specific bind SR/RS dipeptide motifs, phosphorylate SR splicing factors in cells[8] and mediate splicing factor redistribution during the cell cycle[9]. Human SRPK1, the most intensive researched member of this family, is located on chromosome 6p21.2-p21.3, which is expressed in many normal tissues, such as testis and pancreas [10]. SRPK1 has been shown to regulate angiogenesis by interacting with SRSF1 (human SR protein, ASF/SF2) and phosphorylating its RS domain [11]. Phosphorylated SRSF1 is indicative of an angiogenic phenotype as it results in proximal splicing and production of angiogenic VEGF isoforms (VEGF-A165) [12, 13]. It is well known GBM is a highly vascularised tumour, the growth of which needs sufficient oxygen and nutrients from the newly formed blood vessels [14]. The growth of blood vessels is regulated by the the balance between angiogenic factor and anti-angiogenic factor. However, this balance has been destroyed in tumor tissue [15]. The rapid growth of tumor tissue leads to tumor cells living in the state of oxygen hunger and thirst (hypoxia) that can prompt hypoxia inducible factor-1 (HIF-1) activity and induce high expression of HIF-1α protein[16]. HIF-1α induces the expression level of its downstream target genes, such as vascular endothelial growth factor VEGF [17].
In a present review, here is no report about the expression of SRPK1 in glioma and its performance under the hypoxic condition. Here, we found that SRPK1 is expressed in glioma and can promote the growth of U251 cells, even if SRPK1 is present only in neurons and not in glia [18]. More interesting SRPK1 and SRSF1 expression in hypoxic condition can be inhibited and knockdown SRPK1 can inhibit cells invasion, migration and sensitivity to chemotherapy drugs. Also it is significantly higher expressed in low-grade gliomas than in high-grade gliomas, suggesting that SRPK1 as a new molecular player contributing to the early treatment of glioma.
Materials and Methods
Cell lines and treatments
Human glia HA, human lung adenocarcinoma cell A549 and glioma cells U251 and U87 were maintained at 37°C in 5% CO2 (normoxic conditions, 20% O2), in DMEM medium(HyClone) supplemented with 10% fetal bovine serum (FBS, HyClone). Hypoxia treatments, cells were cultured in a tri-gas incubator (HERAcell 150i, Thermo Scientific), which was filled with a 1% O2-5% CO2-94% N2 gas mixture, and stored at 37°C [19]. For DDP or TMZ treatment, cells were cultured in medium with 200 μM TMZ or 20ug/ml DDP in 6 orifice plates for 24h and 48h, respectively.
Glioma tissue
The study included 75 patients who underwent primary surgical resection between 2005 and 2011 at the Linyi People's Hospital; all the diagnoses were made based on the Pathology and Genetics Tumors of Central Nervous System [20] by three pathologists. The clinical information including sex and age was obtained from clinical records. This study was admitted by the Moral and Ethical Committee of the Binzhou Medical University.
Western Immunoblotting
Cells were washed in PBS and then collected in RIPA lysis buffer (Beyotime Biotechnology, China) for protein quantification followed by addition of sample buffer and then heat denatured at 95°C for 10 minutes. Protein lysates were separated using 12% SDS-polyacrylamide gel electrophoresis (PAGE) gels and transferred to Immobilon-P polyvinylidene fluoride (PVDF) membranes (Beyotime Biotechnology). Membranes was blocked with 5% dry non-fat milk, followed by incubation with rabbit Abs against SRPK1 antibody (1:1000, BD Biosciences), mouse anti-SF2/ASF (1:500, SRSF1, Invitrogen), rabbit anti-GAPDH (1:300, Boster Biotechnology), overnight at 4°C. After TBST washing, PVDF membranes were incubated with HRP-conjugated anti-rabbit (Beyotime Biotechnology) or anti- mouse IgG Abs (Boster Biotechnology) for 1h at room temperature (22°C). The immune complexes were visualized by enhanced chemiluminescence using an ECL kit (Beyotime), and detected using ChemiScope3400 Mini (Clinx Science Instruments Co., Ltd, China).
Immunohistochemistry and Immunofluorescence
For immunohistochemical detection of SRPK1 (1:100, Santa Cruz) and CD31 (1:50, Boster), tissue sections were routinely processed and subjected to antigen retrieval. Specimens were then blocked and further incubated with a rabbit monoclonal antibody raised against human SRPK1 or CD31 at a final dilution of 1:200 at 4°C for 12h. The reaction was developed using a DBA chromogenic kit (Boster) at room temperature, and Harris hematoxylin for nuclear staining. Scoring was as follows: 0 (negative), 1+ ( <10-25% of cells weakly positive), 2+ ( >25% of cells weakly positive or <10-25% of cells strongly positive), and 3+ (>25% of cells strongly positive). Cells were fixed with 4% paraformaldehyde 40min at 4°C, washed, permeabilized with 3%TritonX 100 for 20min, incubated with rabbit anti-SRPK1(1:100, Santa Cruz) and mouse anti-SRSF1(1:200, Invitrogen) antibodies for 2h, and mounted in 1:50 goat anti-rabbit TIRTC and goat anti-mouse FITC (Boster Biotechnology) in mounting medium. Images were acquired with a DBI 4000B microscope (Leica)at the Imaging LAS V4.0.
Migration and invasion assays
U251 and U87 cells of nontransfected group, SRPK1 siRNA transfected group (siSRPK1), and siRNA-C (control) transfected group were detached from culture plates in the absence of trypsin (HyClone). Cells were resuspended at a density of 2 × 105/ml in DMEM, 200 μl of the cell suspension was added to the upper chamber of an 8 μm pore size Transwell insert (Corning) in triplicate. DMEM culture solution (600 μl) containing 10% FBS was added to the lower chamber of each well and incubated for 24 h at 37°C. Removed the nonmigratory cells on the upper surface of the membrane and stained cells in 0.1% crystal violet. To count the migrated cells in five random high-power fields, invasion assays were carried out in a similar manner to migration assays. Transwell inserts with 8-μm pores (Corning) were coated with 100 μl Matrigel (Becton Dickinson), which was diluted 1:4 in ice-cold DMEM, and allowed to gel at 37°C, resuspended cells in DMEM and 200ul 5 × 105 /ml cells were seeded in the upper chamber. Culture plates were incubated for 48 h at 37°C, and the cells were stained, and counted as described above.
Quantitative real-time RT-PCR
Total mRNA was extracted by RNAiso Plus (Takara Biotechnology) and 2 ug of mRNA was reverse transcribed using Reverse Transcriptase M-MLV (RNase H-) (Takara). PCRs were performed with the cDNA, specific primers (Takara) and PCR master mix (Takara). The cDNA products were separated by electrophoresis on 1.5% (w/v) agarose gels, visualised under UV following ethidium bromide staining. Quantitative real-time PCR (qRT-PCR) was performed on a iQTM5 Real-Time PCR Optical System (Bio-Rad, USA) with the software version 2.1 following the cycling protocol (at 95°C f or 30 seconds, followed by 40 cycles of 95°C for 10 seconds, and 58°C for 5 seconds). Fluorescence readings were taken during the 60°C step. Measured using SYBR green(Takara) with the following primers: SRPK1 5'-TTCCTCAACTGTAGGTCAGTCATTC-3'(forward, 5'-TGTTCTTGCTCTTGTTCATCTTCAC-3' (reverse), and SRSF1 5'-TTCCTGCCCCAACCAAAC-3' (forward), 5'-AAGGGCTCCAATCGTCAAAA-3' (reverse). Quantitaive real-time PCR was carried out at least 3 times, and a no-template control was included as a negative control.
MTT test, Cell cycle and apoptosis flow cytometry
The cells were firstly seeded in 96-well plates and incubated in the culture medium for 12 h. Next, the cells were exposed to increasing concentrations of TMZ (15-500uM, Xiya Reagent Co. China) or DDP(2.5-80ug/ml, Jinan Qilu-pharma. China) for 24h, 48h or 72 h. Thereafter, the cells were incubated in the culture medium with 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) solution at the final concentration of 0.5 mg/ml for 4h to allow the conversion of MTT into formazan. Then the medium was replaced with dimethyl sulfoxide and absorbance was read at 570 nm using Elisa Bio-Rad Microplate Reader. Cell apoptosis was finally evaluated using the Annexin V-FITC/PI apoptosis detection kit (KeyGEN, China) and cell cycle kit (KeyGEN, China) according to the manufacturer's instructions. Flow cytometry analyses were also performed on Epics XL (Beckman Coulter) and processed using its software.
SRPK1 knockdown in glioma cells
A549, U87 and U251 cells were transfected with siRNA targeting SRPK1 (siRNA438, siRNA716, siRNA1132, Shanghai GenePharma Co.,Ltd) and silencer siRNAs for SRPK1 were selected. The scrambled nontargeting siRNA (GenePharma) was used as a control,. All transfections were carried out using Xfect siRNA transfection reagent (Clontech) according to the manufacturer's instructions. Final concentrations of 300 and 400 pmol siRNA were used in 6-well plate. In all experiments levels of knockdown by RNAi were assessed at the RNA and protein level by PCR and immunoblotting.
Statistical analyses
SPSS16.0 statistical analysis software was used to analyze the experimental data. Student's t-test was used to compare data between two groups (two-tailed, unequal variance). Analysis of variance was conducted to compare data of more than two groups. Chi-square test and one-way analysis of variance (OANOVA). The significance of differences was considered significant when P<0.05 and highly significant when P<0.005.
Results
Overexpressing SRPK1 and SRSF1 in Glioma cells
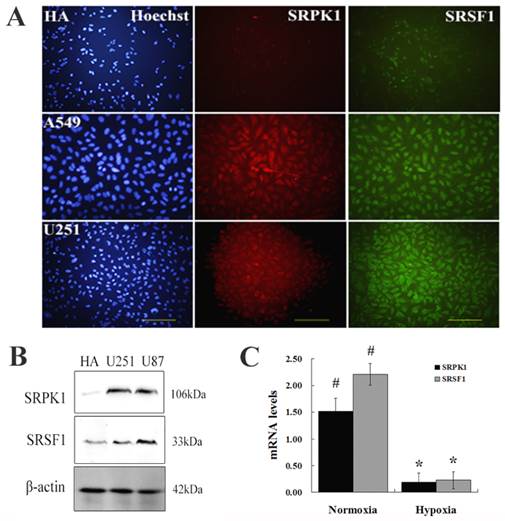
To investigate whether SRPK1 expression in the glioma cells, we chose HA, A549, U251 cells as negative control, positive control and experiment specimens, respectively. Tissue immunofluorescence was carried out for localization the expression of SRPK1 and SRSF1 (Fig.1A). The results showed us that SRSF1 was expressed in glioma nucleus and SRPK1 expressed in glioma cytoplasm and nucleus, and there was a significantly different expression levels of SRPK1 between normal glial and glioma cells by Western bloting (P<0.01) (Fig.1B). Also we found that the expression of SRPK1 and SRSF1 were lower in hypoxic condition than in normoxic condition (Fig.1C). So abnormal expression of SRPK1 and SRSF1 in gliomas may have some roles in cell physiological function.
Overexpression of SRPK1 and SRSF1 in glioma cells. (A) Observed HA, U251 and A549 under fluorescence microscope by Hoechst staining of nuclei (blue), immunostaining for SRPK1 (red) and SRSF1 (green), scale bars: 100μm. (B) Western blot results showed the levels of SRPK1 and SRSF1 in U251, U87 and HA cells, β-Actin was used as an internal loading control. (C) Bar graph showing summary analyses of SRPK1 and SRSF1 with U251 cells by qRT-PCR (# and * P<0.05, n=3, ANOVA) in normoxic and hypoxic condition, compared with HA cells respectively.
SRPK1 influences glioma cells growth in vitro
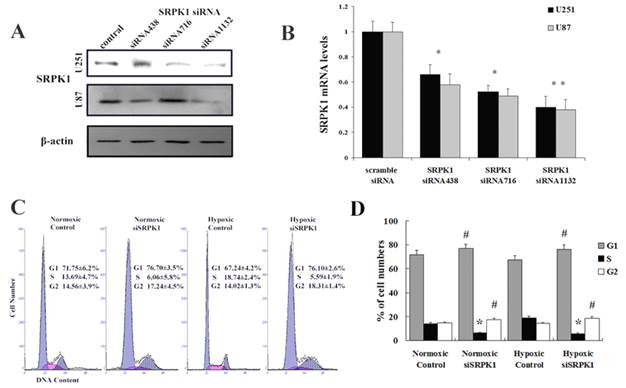
We designed and synthetized siRNA to target SRPK1 (Fig.2A and 2B) and studied whether knockdown SRPK1 influences the growth of glioma cells by flow cytometry. We found that targeting SRPK1 by siRNA had an obvious effect on the cells proliferation in vitro (Fig. 2C). Cell cycle showed the rate of U251 and U87 cells in G1 and G2 phase were significantly higher in the siSRPK1 transfected group than that in the control group (P<0.01) in normoxia and hypoxia (Fig.2D). The rate of glioma cells in S phase was significantly lower than that in the control group (P<0.01) in normoxia and hypoxia (Fig.2C). These results suggest that inhibition of SRPK1 expression can reduce the activity of glioma cells proliferation by causing G0-G1 and G2/M phase arrest either in normoxia or in hypoxia.
Targeting SRPK1 inhibits tumor growth in vitro. (A) Western blot results showed the protein levels of SRPK1 in U251 and U87 cells after 48h transfected with siRNA and siRNA1132 was the best one, β-Actin was used as an internal loading control. (B) RT-PCR detected the mRNA levels of SRPK1 in U251 and U87 cells after 24h transfected with siRNA and siRNA1132 was also the best one, * P<0.05, ** P<0.01. (C,D) The effect of SRPK1 on cell cycle profile was examined by flow cytometric analysis under normal or hypoxic oxygen. G0-G1 and G2-M cell cycle arrest were suggested in SRPK1 siRNA transfected U251 or U87 cells. # and * P<0.05.
SRPK1 can enhance the migration and invasion of glioma cells in normoxic condition
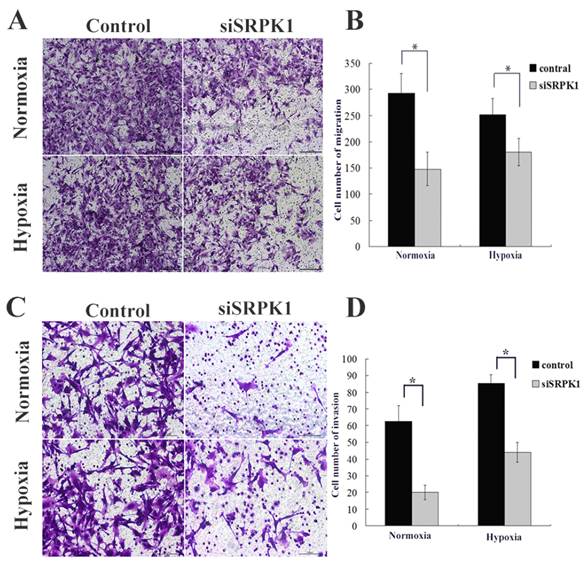
After SRPK1 gene had been inhibited, the migration capacity of U251 and U87 declined, with the cell population coming through the artificial membrane being less than that of the non-transfected group in normoxia (145.3±31.5 vs. 292±38.1, P<0.01, Fig.3A and 3B) and in hypoxia (150.1±25.9 vs. 251.3±31.6, P<0.01, Fig.3A and 3B). No statistical difference in trans-membrane cells was found between the negative control group and the non-transfected group in normoxia (257.7±36.4 vs. 292±38.1, P>0.05, not show) or in hypoxia (236.8±29.8 vs. 251.3±31.6, P>0.05, data not show). The tumor cell invasion through the ECM is an important step in tumor metastasis and we used Matrigel serves as a reconstituted basement membrane matrix. The number of cells migrating to Matrigel was counted. Compared with non-transfected cells, SRPK1 silencing cells decreased invasion significantly in normoxia (20±4.3 vs. 62.8±9.1, P < 0.01, Fig.3C and 3D) and in hypoxia (44±6.1 vs. 85.3±6.4, P<0.01, Fig.3C and 3D). No statistical difference was found between the negative control group and the non-transfected group in normoxia (58.4±6.2 vs. 62.8±9.1, P>0.05, not show) or in hypoxia (79.3±5.8 vs. 85.3±6.4, P>0.05, not show). We found that the invasive ability would be receded in hypoxia compared with in normoxic condition (P<0.05). These results implicate that inhibition of SRPK1 expression can bring down the migration and invasion capacity of glioma cells in normoxia and hypoxia, but the invasion can be restored in hypoxic condition.
Targeting SRPK1 inhibits tumor migration and invasion in vitro. (A) Crystal violet staining of glioma cells that crossed the polycarbonate membrane of the Transwell chamber to detect the migration of cells (100× magnification). (B) Number of cells that crossed the Transwell migration chamber in normoxic and hypoxic condition, * P<0.01. (C) Crystal violet staining of glioma cells that crossed the Matrigel coated polycarbonate membrane of the Transwell chamber to detect the invasion of cells (200× magnification). (D) Number of cells that crossed the Transwell invasion chamber in normoxic and hypoxic condition, * P<0.01.
Inhibition of the SRPK1 gene of U251 cells induces Cisplatin resistance in normoxic and hypoxic condition
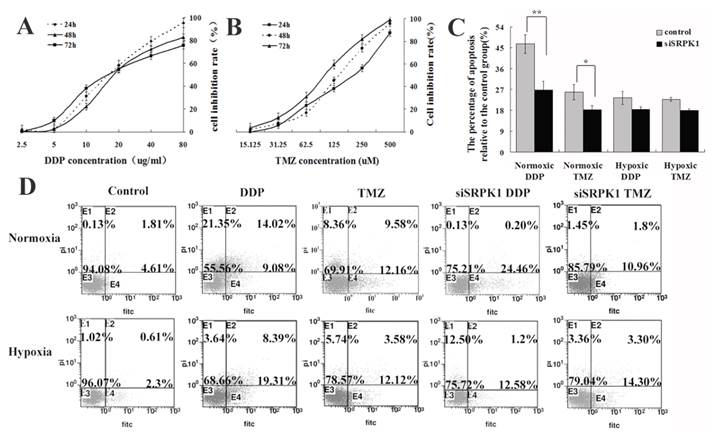
Some researchers have used ovarian, pancreatic carcinoma and male germ cell tumors as a model to elucidate downregulation of the SRPK1 protein conferred resistance to platinum drugs [21,22,23], but in colon and breast carcinoma were reversely[24]. These data indicate that SRPK1 is a cisplatin-sensitive gene, which might potentially play a role in glioma cells. To further evaluate temozolomide(TMZ) effected on the scramble or siSRPK1 U251 cells and investigated cisplatin(DDP) in comparison with TMZ through flow cytometry (Fig.4D). In order to quantify the magnitude of SRPK1's ability to induce chemosensitivity, median lethal dose of DDP and TMZ was firstly identified. A range of 0-80 ug/ml of DDP was determined by MTT and half of cell viability was measured at a concentration of 20 ug/ml (Fig.4A). Similarly, 0-500 uM of TMZ was tested for its effects on half of U251 cell viability and the half lethal concentration was 125 uM of TMZ (Fig.4B). Furthermore, siSRPK1 was added 24h prior to addition of the chemotherapeutics, and the results showed that the inhibited SRPK1 expression could decrease the cells apoposis caused by DDP and TMZ in normoxic condition (P<0.05, Fig.4C and 4D). Interestingly, this role was reversed in hypoxic condition (P>0.05), especially for cells treated by DDP (Fig.4C and 4D).
Downregulated SRPK1 increases chemotherapeutic drug resistance of glioma cells in normoxic and hypoxic condition. (A) Viability of U251 cells exposed to different concentrations of DDP for 24,48,72 h. The results are presented as percentage (mean±SD). Cell viability was evaluated by MTT assay. (B) Viability of U251 cells exposed to different concentrations of TMZ for 24,48,72 h. The results are presented as percentage (mean±SD). Cell viability was evaluated by MTT assay. (C) Bar graph showing the different of cell apoptotic rates between the control and siSRPK1 (* P<0.05, ** P<0.01, n=3, ANOVA) U251 cells treated by DDP or TMZ in normoxic or hypoxic condition(compared with no treatment). (D) U251 cells were transfected for 24h with scramble SRPK1 siRNAs and treated or not for an additional 48 h with 200 uM TMZ or 20ug/ml DDP. Cell apoptotic rates were analyzed by flow cytometry.
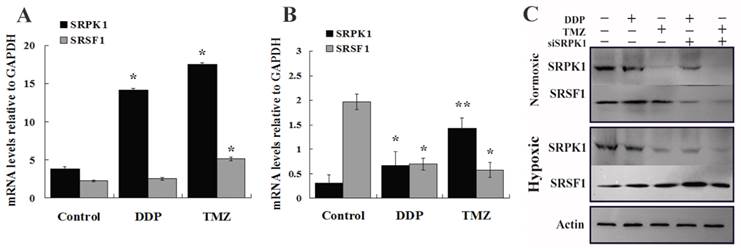
To further validate SRPK1 impacts on the sensitivity of drugs in glioma cells, we studied the changes of SRPK1 and SRSF1 after cells treated by DDP and TMZ in normoxic and hypoxic condition at mRNA and protein levels. RT-PCR analysis was performed to detect the up-regulated SRPK1 mRNA levels that are presented in normoxia after DDP and TMZ treatment (Fig.5A). SRSF1 mRNA was increased by TMZ treatment more than DDP treatment in normoxic condition (Fig.5A). However SRPK1 mRNA levels were mildly elevated, but SRSF1 gene transcription was still inhibited by TMZ treatment in hypoxia (Fig.5B). Western blot results showed that DDP treated cells could promote the release of SRPK1 and SRSF1 protein in nomoxia, while SRPK1 expression was inhibited and the expression of SRSF1 was increased in hypoxia (Fig.5C). SRPK1 was obvious inhibited by TMZ treatment in nomoxia or hypoxia and had little impacts on the whole expression of SRSF1 (Fig.5C). Therefore, SRPK1 can be regarded as a cisplatin-sensitive gene but not as a temozolomide -sensitive gene.
To explicit SRPK1 can increase DDP resistance of glioma cells in molecular levels. (A) The relative expression of SRPK1 and SRSF1 mRNAs in U251 cells treated by DDP or TMZ compared with control group in normoxic condition (*P<0.05). (B) RT-PCR detected SRPK1 and SRSF1 of U251 cells treated by DDP or TMZ compared with control group in hypoxic condition (*P<0.05, ** P<0.01). (C) The picture showed the expression of SRPK1 and SRSF1 in U251 cells by Western blot after 24h transfected with siRNA and treated or not with TMZ or DDP, β-Actin was used as an internal loading control.
SRPK1 has different expression levels in different classification of Glioma tissues
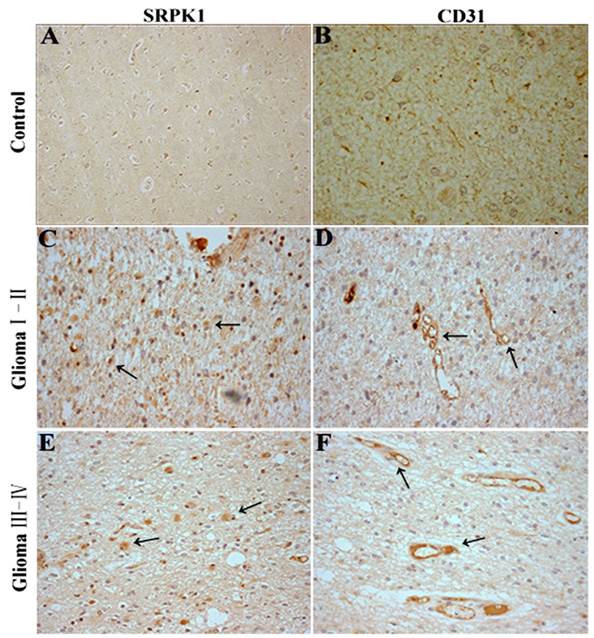
Recent studies have identified SRSF1 is phosphorylated by SRPK1 [25] and nuclear SRSF1 can regulate proximal splicing of angiogenic VEGF isoforms [12].To further assess if SRPK1 protein expression has a relationship with glioma angiogenesis, we performed SRPK1 and CD31 IHC on 75 clinical pathological specimens from patients with primary glioma. The results showed SRPK1 was weakly expressed in neuron at the adjacent normal tissue of glioma (Fig.6A), and little CD31 staining was observed (Fig.6B). However, SRPK1 and CD31 were strongly expressed in the different gliomas (Fig.6C-6F). SRPK1 was significantly upregulated in 62.67% (47 of 75) of glioma tissues, and was more expressed in low-grade (I,II) gliomas than in high-grade (III,IV) gliomas (Table 1). There is no significant difference in sexuality and age from grade I to IV (P>0.05, Table 1). Also, we found that SRPK1 had a more obvious positive expression around the vascular area than away from the vascular area (85±8.6 vs. 49.3±6.9, P<0.05). In summary, consistent with our prior analysis, SRPK1 is significantly upregulated in glioma tissues and expressed more in low-grade gliomas than in high-grade gliomas. SRPK1 expression may affect the prognosis of patients glioma. Furthermore, SRPK1 is more distributed in the surrounding blood vessels and this kind of phenomenon maybe more conducive to promoting glioma angiogenesis.
SRPK1 expression in glioma tissue with different grades.
| Cases | SRPK1 protein expression (n) | P value | |||
|---|---|---|---|---|---|
| Negative | Weak | Strong | |||
| (75) | (28) | (25) | (22) | ||
| Sex | P = 0.655 | ||||
| Female | 35 | 14 | 11 | 10 | |
| Male | 40 | 14 | 14 | 12 | |
| Age | P = 0.922 | ||||
| ≤ 59 Y | 45 | 17 | 16 | 12 | |
| ≥ 60 Y | 30 | 11 | 9 | 10 | |
| WHO classification of glioma | P = 0.004* | ||||
| Grade I | 8 | 1 | 3 | 4 | |
| Grade II | 19 | 3 | 6 | 10 | |
| Grade III | 25 | 11 | 9 | 5 | |
| Grade IV | 23 | 12 | 7 | 4 | |
* Compared with Grade I-II and Grade III-IV, the low-grade gliomas have more positive expression of SRPK1 than high-grade gliomas.
SRPK1 and CD31 immunohistochemistry(IHC) in glioma tissues. (A, B) SRPK1 and CD31 IHC staining in normal brain tissues around the excision of tumor tissue. (C, D) IHC staining for SRPK1 and CD31 staining in glioma I-II tissues. (E, F) IHC staining for SRPK1 and CD31 staining in glioma III-IV tissues. All photomicrographs were taken at 200× magnification; haematoxylin counterstain. Arrows indicate the positive expression areas.
Discussion
In this study, we the first time describe the knockdown SRPK efficiently can inhibit the growth, migration invasion and chemosensitivity of glioma cells in normoxic and hypoxic condition.
SRPK1 can regulate the activity of RS proteins to control cell-cycle at G2/M phase mainly in vitro [12,26], although our findings are generally consistent with these studies, but glioma cells arrest in G0/G1 phase is significantly increased. Maybe siSRPK1 affects the activity of SRPK2 (one of SR/RS protein kinases family) which can bind and phosphorylate acinus, low expression SRPK2 arrests cells at the G1 phase [27]. Some studies also show that SRPK1 can through Akt phosphorylation to link with PI3K/Akt signaling, accordingly affect the cells proliferation [28]. And there is no significant different after transfected siSRPK1 between at normoxic or hypoxic condition, even if in hypoxic conditions can promote the synthesis of HIF-1α which contributes to tumor cell survival[29] and activation of oncogenic pathways (PI3K/Akt) in cancer cells could result in an increased expression of HIF-1α[30]. In addition, GBMs are characterized by a high degree of migration and invasion [3, 31]. We controlled the expression of SRPK1 by siRNA, the results showed that SRPK1 low expression in glioma cells could distinctly reduce their invasion and migration capability no matter in normoxia or hypoxia. In spite of there has no any studies about SRPK1 impacts on tumor migration and invasion. It is interesting that hypoxia can reduce SRPK1 knockdown-induced tumor cell migration and invasion ability. Jeon et al demonstrated that hypoxia induced accumulation of HIF-1α which could augment levels of MMP-2, MMP-9 or uPA [32, 33] through the MAPK signalling pathway [34] to affect tumour angiogenesis, metastasis and growth factor release from the extra-cellular matrix (ECM) [35]. So we consider that SRPK1 may be through MAPK [14] or PI3K/Akt signaling pathway to play corresponding roles in metastasis and this influence can be turned by hypoxia. Therefore this will become a new research hotspot.
In the present review, tumor hypoxia present a highly malignant phenotype and is associated with resistance to many chemotherapeutic drugs[36], which limits the ability of drugs to penetrate tumor tissue in a potentially lethal concentration[37]. Also hypoxia induces the activation of VEGF gene transcription through an HIF-dependent mechanism[38] and the high level of VEGF is believed to be responsible for the glomeruloid microvascular proliferation that is characteristic of GBM[39] leads to tumor growth and angiogenesis. Meanwhile, the activation of SRPK1 affects the chemotherapy sensitivity of tumor cells, such as enhance cisplatin sensitivity in retinoblastoma and ovarian carcinoma [40,41], but can be conversely in colonic and pancreatic carcinomas[14]. And SRSF1 can be phosphorylated by SRPK1 to monitor the different expression of VEGF as well. SRPK1 plays a major role in normoxic conditions, whereas in a hypoxic environment, inhibition of SRPK1 and its downstream proteins expression may be caused by the low ATP/ADP exchange rates [42]. Furthermore, SRPK1 is regarded as platinum drugs sensitivity gene in normoxia and can be abolished in hypoxia, hypoxic cells present more resistant to cisplatin in some cell lines [43] but also showing cisplatin to be a HIF-1 inhibitor[44] that can encourage SRPK1 expression. But TMZ as a kind of new alkylating agent can clearly inhibit SRPK1 expression in protein level, so SRPK1 is not regarded as TMZ drugs sensitivity gene even if siSRPK1 can slightly effect of cells apoposis.
In summary, SRPK1 has more performance at low-grade glioma that can furnish sufficient blood to tumors against hypoxia and maintain sensitivity to chemotherapeutic drugs. With the elevated of tumor grade, the roles of SRPK1 are diminishing and the roles of HIF-1 are gradually appeared in hypoxic situation. In addition, HIF-1α is primarily localized in pseudopalisading cells around areas of necrosis[45],though we can find new blood vessels around of HIF-1α and SRPK1, they have some different parts of glioma tissues. So we need to further discussion the relationship between SRPK1 and HIF-1, and find more precise theoretical basis for the treatment of gliomas.
Acknowledgements
This study was financially supported by the Key Project of National Natural Science Foundation of Shandong Province (ZR 2009CL004), China Postdoctoral Science Foundation (20100481466), Pharmaceutical Health Science and Technology Development Program of Shandong Province (2011QZ001) and National Natural Science Foundation of China (81171142/H0910).
Conflict of Interest
No potential conflicts of interest were disclosed.
References
1. Louis DN. Molecular pathology of malignant gliomas. Annu Rev Pathol Mech Dis. 2006;1:97-117
2. Dietrich J, Diamond EL, Kesari S. Glioma stem cell signaling: therapeutic opportunities and challenges. Expert Rev Anticancer Ther. 2010;10(5):709-722
3. Louis DN, Ohgaki H, Wiestler OD. et al. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007;114:97-109
4. Wrensch M, Minn Y, Chew T. et al. Epidemiology of primary brain tumors: current concepts and review of the literature. Neuro-oncol. 2002;4:278-299
5. Stupp R, Mason W, van den Bent MJ. et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987-996
6. Elizabeth A, Maher EA, Furnari FB. et al. Malignant glioma: genetics and biology of a grave matter. Genes. 2001;15:1311-1333
7. Gui JF, Lane WS, Fu XD. A serine kinase regulates intracellular localization of splicing factors in the cell cycle. Nature. 1994;369:678-682
8. Hayes GM, Carrigan PE, Miller LJ. Serine-arginine protein kinase 1 overexpression is associated with tumorigenic imbalance in mitogen-activated protein kinase pathways in breast, colonic, and pancreatic carcinomas. Cancer Res. 2007;67:2072-2080
9. Wang HY, Lin W, Dyck JA. et al. SRPK2: a differentially expressed SR protein-specific kinase involved in mediating the interaction and localization of pre-mRNA splicing factors in mammalian cells. J. Cell Biol. 1998;140:737-750
10. Papoutsopoulou S, Nikolakaki E, Chalepakis G. et al. SR protein-specific kinase 1 is highly expressed in testis and phosphorylates protamine 1. Nucleic Acids Res. 1999;27:2972-80
11. Aubol B.E, Chakrabarti S, Ngo J. et al. Processive phosphorylation of alternative splicing factor/splicing factor 2. Proc Natl Acad Sci USA. 2003;100:12601-12606
12. Nowak DG, Amin EM, Rennel ES. et al. Regulation of vascular endothelial growth factor (VEGF) splicing from pro-angiogenic to anti-angiogenic isoforms: a novel therapeutic strategy for angiogenesis. J Biol Chem. 2010;285(8):5532-40
13. Amin EM, Oltean S, Hua J. et al. WT1 mutants reveal SRPK1 to be a downstream angiogenesis target by altering VEGF splicing. Cancer Cell. 2011;20(6):768-80
14. Parkin DM, Bray F, Ferlay J. et al. Global cancer statistics, 2002. CA Cancer J Clin. 2005;55(2):74-108
15. Carmeliet P, Jain RK. Angiogenesis in cancer and other diseases. Nature. 2000;407:249-257
16. Hicklin DJ, Ellis LM. Role of the vascular endothelial growth factor pathway in tumor growth and angiogenesis. J Clin Oncol. 2005;23:1011-1027
17. Cassavaugh J, Lounsbury KM. Hypoxia-mediated biological control. J.Cell Biochem. 2011;112:735-744
18. Harper SJ, Bates DO. VEGF-A splicing: the key to anti-angiogenic therapeutics? Nat Rev Cancer. 2008;8(11):880-887
19. Mytilinaios DG, Tsamis KI, Nikolakaki E. et al. Distribution of SRPK1 in human brain. J Chem Neuroanat. 2012;43(1):20-7
20. Oda T, Hirota K, Nishi K. et al. Activation of hypoxia-inducible factor 1 during macrophage differentiation. Am J Physiol Cell Physiol. 2006;291(1):C104-13
21. Cavanee WK. WHO classification of tumours: Pathology and Genetics of Tumours of the Nervous System. Lyon: IARC Press. 2000
22. Burger H, Capello A, Schenk PW. et al. A genome-wide screening in Saccharomyces cerevisiae for genes that confer resistance to the anticancer agent cisplatin. Biochem Biophys Res Commun. 2000;269:767-774
23. Schenk PW, Boersma AWM, Brandsma JA. et al. SKY1 is involved in cisplatin-induced cell kill in Saccharomyces cerevisiae and inactivation of its human homologue, SRPK1, induces cisplatin resistance in a human ovarian carcinoma cell line. Cancer Res. 2001;61:6982-6986
24. Schenk PW, Brok M, Boersma AWM. et al. Anticancer drug resistance induced by disruption of the Saccharomyces cerevisiae NPR2 gene: a novel component involved in cisplatin- and doxorubicin-provoked cell kill. Mol Pharmacol. 2003;64:259-268
25. Plasencia C, Martínez-Balibrea E, Martinez-Cardús A. et al. Expression analysis of genes involved in oxaliplatin response and development of oxaliplatin-resistant HT29 colon cancer cells. Int J Oncol. 2006;29(1):225-35
26. Zhong X.Y, Ding J.H, Adams J.A. et al. Regulation of SR protein phosphorylation and alternative splicing by modulating kinetic interactions of SRPK1 with molecular chaperones. Genes Dev. 2009;23:482-495
27. Ding JH, Zhong XY, Hagopian JC. et al. Regulated cellular partitioning of SR protein-specific kinases in mammalian cells. Mol Biol Cell. 2006;17(2):876-85
28. Jang SW, Yang SJ, Ehle´n A. et al. Serine/arginine protein-specific kinase 2 promotes leukemia cell proliferation by phosphorylating acinus and regulating cyclin A1. Cancer Res. 2008;68:4559-4570
29. Bo Zhou, Yandong Li, Qing Deng. et al. Han SRPK1 contributes to malignancy of hepatocellular carcinoma through a possible mechanism involving PI3K/Akt. Mol Cell Biochem. 2013;379:191-199
30. Hirota K, Semenza GL. Regulation of angiogenesis by hypoxia -inducible factor 1. Crit Rev Oncol Hematol. 2006;59:15-26
31. Ke Q, Costa M. Hypoxia-inducible factor -1 (HIF -1). Mol Pharmacol. 2006;70(5):1469-1480
32. Wu M, Chen Q, Li D. et al. LRRC4 inhibits human glioblastoma cells proliferation, invasion, and proMMP-2 activation by reducing SDF-1 alpha/CXCR4-mediated ERK1/2 and Akt signaling pathways. J Cell Biochem. 2008;103:245-255
33. Jeon HW, Lee KJ, Lee SH. et al. Attenuated expression and function of the RECK tumor suppressor under hypoxic conditions is mediated by the MAPK signaling pathways. Arch Pharm Res. 2011;34(1):137-45
34. Park SY, Nho CW, Kwon DY. et al. Maslinic acid inhibits the metastatic capacity of DU145 human prostate cancer cells: possible mediation via hypoxia-inducible factor-1α signalling. Br J Nutr. 2013;109(2):210-22
35. Soeda A, Park M, Lee D. et al. Hypoxia promotes expansion of the CD133-positive glioma stem cells through activation of HIF-1alpha. Oncogene. 2009;28:3949-3959
36. Mandal SK, Rao LV, Tran TT. et al. A novel mechanism of plasmin-induced mitogenesis in fibroblasts. Journal of Thrombosis and Haemostasis. 2005;3:163-169
37. Vaupel P, Mayer A. Hypoxia in cancer: significance and impact on clinical outcome. Cancer Metastasis Rev. 2007;26:225-239
38. Minchinton AI, Tannock IF. Drug penetration in solid tumours. Nat Rev Cancer. 2006;6(8):583-592
39. Forsythe J.A, Jiang B.H, Iyer N.V. et al. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol. Cell Biol. 1996;16:4604-4613
40. Brat D.J, Van Meir E.G. Vaso-occlusive and prothrombotic mechanisms associated with tumor hypoxia, necrosis, and accelerated growth in glioblastoma. Lab Invest. 2004;84:397-405
41. Dai S, Huang ML, Hsu CY, Chao KS. Inhibition of hypoxia inducible factor 1alpha causes oxygen-independent cytotoxicity and induces p53 independent apoptosis in glioblas-toma cells. Int J Radiat Oncol Biol Phys. 2003;55:1027-1036
42. Krishnakumar S, Mohan A, Kandalam M. et al. SRPK1: a cisplatin sensitive protein expressed in retinoblastoma. Pediatr Blood Cancer. 2008;50(2):402-6
43. Aubol BE, Plocinik RM, McGlone ML. et al. Nucleotide release sequences in the protein kinase SRPK1 accelerate substrate phosphorylation. Biochemistry. 2012;51(33):6584-94
44. Wartenberg M, Ling FC, Muschen M. et al. Regulation of the multidrug resistance transporter P-glycoprotein in multicellular tumor spheroids by hypoxia-inducible factor (HIF-1) and reactive oxygen species. FASEB J. 2003;17(3):503-505
45. Zagzag D, Zhong H, Scalzitti JM. et al. Expression of hypoxia-inducible factor 1alpha in brain tumors: association with angiogenesis, invasion, and progression. Cancer. 2000;88:2606-2618
Author contact
![]() Corresponding author: Fei Huang, PhD, Institute of Human Anatomy and Histology and Embryology, Otology & Neuroscience Center, Binzhou Medical University, 346 Guanhai Road, Laishan, Shandong Province, 264003, China. Tel: 0535-6913076; Fax: 0535-6913077; E-Mail: hfei22518com or Fengchan Han, PhD, Institute of Human Anatomy and Histology and Embryology, Otology & Neuroscience Center, Binzhou Medical University, 346 Guanhai Road, Laishan, Shandong Province, 264003, China. Tel: 0535-6913480; Fax: 0535-6913077; E-Mail: hanfengchancom.
Corresponding author: Fei Huang, PhD, Institute of Human Anatomy and Histology and Embryology, Otology & Neuroscience Center, Binzhou Medical University, 346 Guanhai Road, Laishan, Shandong Province, 264003, China. Tel: 0535-6913076; Fax: 0535-6913077; E-Mail: hfei22518com or Fengchan Han, PhD, Institute of Human Anatomy and Histology and Embryology, Otology & Neuroscience Center, Binzhou Medical University, 346 Guanhai Road, Laishan, Shandong Province, 264003, China. Tel: 0535-6913480; Fax: 0535-6913077; E-Mail: hanfengchancom.