Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2013; 4(8):653-661. doi:10.7150/jca.7626 This issue Cite
Review
Kisspeptin/KISS1R System in Breast Cancer
Donna Cvetković1, Andy V. Babwah 1,2,3,4, Moshmi Bhattacharya1, 3, 5 ![]()
1. Department of Physiology and Pharmacology, Schulich School of Medicine and Dentistry, University of Western Ontario, London, Ontario, Canada, N6A 5C1;
2. The Children's Health Research Institute, London, Ontario, Canada;
3. Lawson Health Research Institute, London, Ontario, Canada;
4. Department of Obstetrics and Gynaecology, University of Western Ontario London, Ontario, Canada N6C 2V5;
5. Department of Oncology, University of Western Ontario, London, Ontario, Canada.
Received 2013-9-10; Accepted 2013-9-22; Published 2013-9-26
Abstract
Kisspeptins (KP), peptide products of the kisspeptin-1 (KISS1) gene are the endogenous ligands for a G protein-coupled receptor (GPCR) - KP receptor (KISS1R). KISS1R couples to the Gαq/11 signaling pathway. KISS1 is a metastasis suppressor gene and the KP/KISS1R signaling has anti-metastatic and tumor-suppressant effects in numerous human cancers. On the other hand, recent studies indicate that KP/KISS1R pathway plays detrimental roles in breast cancer. In this review, we summarize recent developments in the understanding of the mechanisms regulating KP/KISS1R signaling in breast cancer metastasis.
Keywords: Kisspeptin, KISS1R, breast cancer, metastasis, invasion, G protein-coupled receptor.
Introduction
The metastasis of cancer cells hinges upon a series of successive events; hence, interrupting any one step could halt the process. Metastasis suppressors, defined by their abilities to inhibit metastasis without blocking orthotopic tumor growth, are attractive agents to treat metastasis[1]. Over a decade ago, a new metastasis suppressor gene was identified and named KISS1 in reference to its place of discovery - Hershey, Pennsylvania, the home of the famous Hershey Kisses[2]. The KISS1 gene encodes a 145-amino acid protein, which is subsequently cleaved into a 54-amino acid fragment (named metastin), this in turn is cleaved into even shorter, biologically active peptides (10, 13, 14 amino acids long), collectively referred to as kisspeptins (KPs)[3-5]. KP-10 is the smallest active peptide comprised of the last ten amino acids of the full 145-amino acid peptide[6]. In humans, reverse transcription polymerase chain reaction revealed KISS1 mRNA to be expressed at high levels in the brain, breast, pancreas, placenta, testis, liver, heart and small intestine[5, 7-9].
Although the sequence for the KISS1 gene has been known since its initial discovery, it was not until 2001 that the peptide products of the KISS1 gene were identified as the endogenous ligands for the KP receptor (KISS1R; formerly known as AXOR12, GPR54 or hOT7T175) by three independent groups[3, 5, 8]. KISS1R mRNA displays similar tissue distribution as its ligand, with high levels expressed in the placenta, pituitary gland, pancreas, breast and spinal cord[3, 5, 8, 9]. KISS1R mRNA is also present in the heart, skeletal muscle, kidney, liver, placenta and central nervous system[3, 8, 10]. Radioligand binding of 125I-KP-14 was detected in aorta, coronary artery and umbilical vein[4], suggesting expression of the KISS1R in the vasculature.
Kisspeptin/KISS1R Signaling
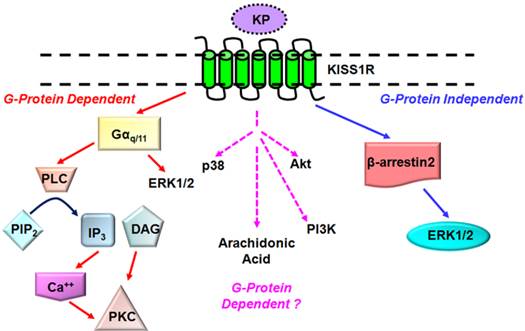
KISS1R is a G protein-coupled receptor (GPCR) that shares high sequence identity with the galanin receptor[3, 5, 8]. KISS1R couples to the Gαq/11 signaling pathway and activates the primary effector phospholipase C (PLC). Activation of the PLC results in the formation of inositol-(1,4,5)-trisphosphate (IP3) and diacylglycerol, mobilization of calcium and activation of protein kinase C (PKC) and extracellular signal-regulated kinases 1 and 2 (ERK1/2)[11-13] (Fig. 1). These secondary messengers are associated with KISS1R signaling in a variety of cell types and may be ubiquitous components of the KP/KISS1R signaling system[3, 5, 8]. Other secondary effectors of the KP/KISS1R signaling pathway which may prove to regulate KISS1R signaling in a cell-specific manner include p38, phosphatidylinositol-3-kinase (PI3K), Akt and arachidonic acid[3, 8, 14, 15] (Fig. 1). Notably, emerging findings are showing that KISS1R activates a plethora of signaling molecules that have the potential to regulate a diverse array of biological processes, and that is in fact being observed and will be discussed later.
Established and putative signaling pathways coupled to KISS1R. KISS1R is a Gq/11-coupled receptor, resulting in the activation of the primary effector phospholipase C (PLC) and associated secondary effectors such as protein kinase C (PKC) and ERK1/2. KISS1R can also signal via G protein-independent (β-arrestin-dependent) pathway to also activate ERK1/2. KISS1R activates several secondary effectors including p38, phosphatidylinositol-3-kinase (PI3K) but it is not yet known through which pathway they are activated.
One of the principal mechanisms for switching off GPCR signaling is homologous desensitization, a process that involves the co-ordinated actions of two families of proteins, the GPCR kinases (GRKs) and β-arrestins[16, 17]. For the prototypical GPCR, such as the α-adrenergic receptor, the ligand-bound receptor undergoes GRK-dependent phosphorylation at residues on its intracellular loops and carboxyl terminus. Phosphorylation of the receptor promotes the high-affinity binding of β-arrestins-1 and -2 to the receptor, which physically blocks further coupling to G proteins[16, 17]. β-arrestins also target many GPCRs for internalization in clathrin-coated vesicles; once internalized, the receptor is either directed for degradation via lysosomes or recycling back to the cell surface via recycling endosomes[16, 17].
KISS1R has been shown to be constitutively associated with GRK2 and β-arrestins-1 and -2, and as noted for many GPCRs, these interactions are mediated through residues in the second intracellular loop and cytoplasmic tail of KISS1R[12]. KISS1R undergoes GRK2-dependent desensitization, however, this occurs in a phosphorylation independent manner[11]. The G protein-uncoupled receptor then undergoes β-arrestin-dependent internalization via clathrin-coated pits[12]. Recent studies have shown that the bulk of internalized KISS1R is rapidly recycled back to the cell surface with little being targeted for degradation[18]. This led to the recognition that through rapid recycling, the cell maintains an active pool of KISS1R at the cell surface permitting prolonged KP-dependent signaling in the presence of continuous receptor desensitization[11]. This finding is crucial to the future development of any KP-based therapy.
For a number of GPCRs, β-arrestins also act as molecular adapters and recruit signaling proteins to the agonist-occupied GPCR[19]. Thus, in their own right, β-arrestins also act as signaling molecules. Recently, members of our group have demonstrated that KISS1R also undergoes β-arrestin-dependent signaling in several cell types including breast cancer MDA-MB-231 cells[12, 13]. Specifically, in the MDA-MB-231 cell line, they demonstrated that β arrestin-2 mediates a robust activation of ERK1/2[12]. Importantly, it is now demonstrated that KISS1R activates ERK1/2 via both Gαq/11 and β-arrestin-2; hence, ERK1/2 represents a major regulator of the KP/KISS1R signaling[12]. In many human cancers, ERK1/2 activity is dysregulated and this altered activity is often implicated in tumorigenesis and resistance to anticancer therapies[20].
The functional roles of the KP/KISS1R system have been extensively reviewed[9,21]. Physiologically, the KP/KISS1R signaling system potently triggers gonadotropin-releasing hormone release, thereby, acting a major regulator of the neuroendocrine reproductive axis[22, 23]; negatively regulates trophoblast migration and invasion, thereby, being proposed as a major regulator of placentation and fetal development[24]. In less investigated roles, the KP/KISS1R signaling system has also been proposed as a regulator of insulin secretion, cardiovascular function and kidney development (reviewed in[9, 25]). The rest of this review article will focus on the roles of KPs in regulating breast cancer biology. In many cancers, KP/KISS1R signals as a metastasis suppressor. However, we will focus on some controversial findings in breast cancer, where the role of KP/KISS1R has been difficult to discern and discuss the latest findings that shed light on the mechanisms by which KISS1R regulates breast cancer metastasis. Since KISS1R is emerging as a GPCR of widespread clinical importance, understanding the molecular mechanisms that regulate KISS1R signaling upon receptor activation is necessary to evaluate its potential as a therapeutic target.
Roles of KP/KISS1R as a Metastasis Suppressor
To date, the metastasis suppressor activity of KPs have been identified in numerous cancers, including melanoma[2,7,26-28], thyroid[29,30], ovarian[31-34], bladder[35,36], gastric[37,38], esophageal[39], pancreatic[40], lung[41] and pituitary[42] cancers. The anti-cancer roles of KP/KISS1R have been recently discussed in excellent reviews[9,43-45]. For several of these cancers, a unified emerging mechanism underlying KP/KISS1R-dependent metastatic suppression is the repression of matrix metalloproteinase (MMP)-9 activity and subsequent inhibition of cancer cell migration and invasion[44, 46, 47]. Increased activation of focal adhesion kinase (FAK), leading to the formation of excessive focal adhesions and stress fibre formation have also been observed and believed to further contribute to reduced cancer cell motility[3, 48].
Clinically, a reduction in KISS1 and/or KISS1R expression has been shown to be associated with poor prognosis in cancer patients and consequently, the expression of KISS1 and/or KISS1R could potentially prove to be powerful prognostic markers in clinical settings[7, 28, 32, 37-39, 41, 49-56]. Studies have also emerged that indicate that the KP/KISS1R signaling promotes tumorigenesis. For instance, in hepatocellular cancers, overexpression of KISS1 and KISS1R has been observed and correlates with cancer progression[57-59]. Furthermore, KISS1 expression level in hepatocellular carcinoma correlated with worsened clinical outcome, as an independent prognostic marker for the aggressiveness of hepatocellular carcinoma[57-59]. Additionally, KP/KISS1R signaling appears to have detrimental effects in breast cancer.
KP/KISS1R Signaling in Breast Cancer Metastasis
Initially, KISS1 was thought to functions as a metastasis suppressor gene in human breast cancer, similar to its anti-metastatic roles observed in melanoma[60]. However, this study was done using human 'breast' cancer MDA-MB-435 cells, which have been shown to have gene expression profile more closely resembles that of melanoma cell lines, rather than that of other breast tumor cell lines[61]. Thus studies performed using this cell line must be interpreted with caution[62].
A recent landmark study established a role for KP/KISS1R signaling in regulating breast cancer metastasis using a mouse mammary tumor virus-polyoma virus middle T antigen (MMTV-PyMT) model[63]. In this model, the transformation of the mammary epithelium leads to the development of multifocal mammary adenocarcinomas and metastatic lesions in the lymph nodes and in the lungs. Cho and colleagues showed that Kiss1r haploinsufficiency (Kiss1r+/-) delayed PyMT-induced breast tumor initiation, latency, growth and metastasis[63]. Orthotopic injection of isolated mouse primary breast cancer MMTV-PyMT/Kiss1r+/- cells into NOD.SCID/NCr immunocompromised mice showed reduced primary tumor growth, compared to mice injected with MMTV-PyMT/Kiss1r+/+ cells, indicating that reduced expression of the receptor inhibits tumor growth in vivo. Additionally, the authors showed that KISS1R plays a vital role in Ras-induced MCF10A cell tumorigenesis signaling via RhoA GTPase downstream of Gαq activation. Although the authors did not provide any evidence that KISS1R regulates metastasis using a xenograft mouse model, this study clearly establishes that KISS1R positively regulates tumorigenesis and metastasis although the underlying molecular mechanism remain to be deciphered.
KISS1 Expression in Human Breast Tumors
The first evidence that KISS1 expression is elevated in patient breast tumors was shown in a study by Martin and colleagues[64]. They demonstrated that KISS1 mRNA expression was elevated in node-positive tumors, compared to node-negative ones and that KISS1 mRNA and protein expression increased with tumor grade as determined by quantitative-PCR analysis and immunohistochemistry, respectively. Moreover, they found that patients with metastatic disease had elevated levels of KISS1 expression compared to healthy individuals, and that this associated with poor patient prognosis. A rise in KISS1R mRNA expression in node-positive tumors was observed, although this was not statistically significant. This study also showed that the over-expression of KISS1 in MDA-MB-231 human breast cancer cells increased their invasiveness and decreased their adhesive property using Matrigel-coated transwell chamber invasion assays, thus, providing evidence that KISS1 may not be functioning as a metastasis suppressor in breast cancer cells[64].
In sharp contrast to the study by Martin and colleagues[64], other reports indicate that KISS1 mRNA and protein expression is absent in node-positive tumors, and found a significant negative correlation with axillary lymph node involvement[52, 65, 66]. No association of KISS1 expression was found with genes that regulate cell-cycle and proliferation such as HER2, VEGF and p53[52]. KISS1 mRNA and protein expression was also found to be significantly higher in primary breast cancer compared with breast tumors that metastasized to the brain[67, 68]. In fact, a significant down-regulation of KISS1 expression has been reported in brain metastatic lesions, compared to primary ductal carcinomas and this correlated with immunohistochemical analysis of KISS1 protein levels[64]. This suggests that a loss of KISS1 may contribute to the formation of distant metastases, since there was a down-regulation of KISS1 expression in the metastases compared to primary tumors [67]. Thus, these studies support the anti-metastatic role of KISS1[52, 67].
The reason for these contradictory results remain unclear; however, with the analysis of patient samples, factors have to be taken into consideration such as whether the patients had received chemotherapy, the age of the women (pre- versus post-menopausal) and the genetic diversity of the patient population. These factors are not always considered or included as part of the analysis. Interestingly, these later studies did not examine KISS1R association with axillary lymph node involvement and thus, further studies are necessary to better understand changes in the expression profile of KISS1 and KISS1R in patients at the gene and protein level. Furthermore, recent studies have shown that the estrogen receptor (ER) status of breast tumors has to be taken into consideration since expression and function of both KISS1 and KISS1R are modulated by estrogen.
Regulation of KISS1 and KISS1R Expression by Estrogen in Breast Cancer
The steroid hormone estrogen plays a critical role in the development of the mammary epithelium during puberty[69-72]. Estradiol (E2) acting through ERα is required for the normal growth and development of the mammary ductal network[71,73]. ERα activation is mitogenic in normal and transformed cells, but is able to suppress processes such as epithelial-mesenchymal transition (EMT)[69, 73].
It is well established through studies using mice and rats, that in the hypothalamus, KISS1 mRNA level is directly regulated by E2[74, 75]. ERα-mediated pathways play a crucial role in breast carcinogenesis; moreover, ERα is an important prognostic indicator in breast cancer and of the response to endocrine therapy[76, 77], with breast tumors typically categorized as being ER-positive or ER-negative[71]. In a 2007 study, Marot and colleagues investigated whether the expression of KISS1 and KISS1R is regulated by estrogen and whether this is clinically relevant. They found that KISS1 and KISS1R expression was negatively regulated by E2. Furthermore, among post-menopausal women with ERα-positive tumor samples who had been treated with the ER antagonist, tamoxifen, patients with shorter relapse-free survival had elevated expression of KISS1 and KISS1R mRNA compared to tumors expressing low mRNA levels of these genes. They reported that KISS1 mRNA levels were high in breast tumors, compared to normal mammary tissue, whereas KISS1R mRNA levels were high in invasive tumors compared to normal tissue[78]. In contrast to this study, Jarzabek and others reported that ER-positive tumors exhibited higher KISS1 and KISS1R levels than ER-negative tumors[50]. This study also revealed that KISS1R expression is negatively associated with HER2 status in breast cancer[50]. Thus the expression profile and roles of KISS1 and KISS1R in breast cancer has been difficult to discern and this might simply reflect the great heterogeneity among breast tissue, in addition to the fact that mRNA levels may not necessarily correlate to protein levels.
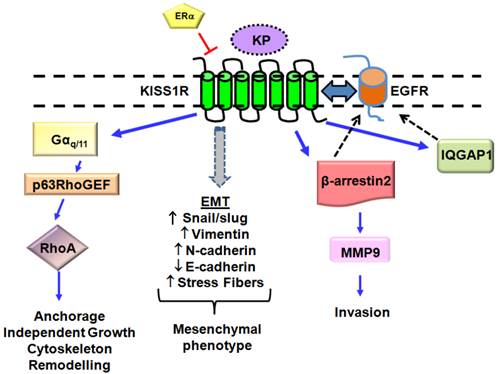
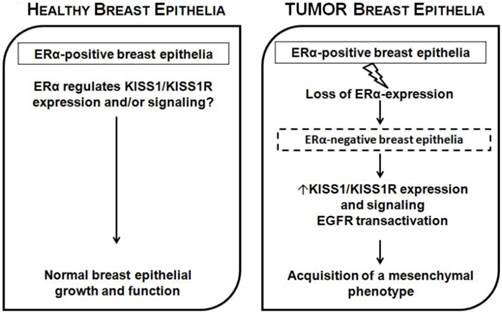
To further study the regulation of KP/KISS1R by ERα, ERα was over-expressed in the ERα-negative MDA-MB-231 breast cancer cells[78, 79]. Treatment with E2-induced a decrease in KISS1 mRNA levels as well as KISS1R protein levels, compared to controls[78,79]. The expression of ERα (in the absence of E2) inhibited KP-10-stimulated cell migration and invasion, and KP-10-stimulated epidermal growth factor receptor (EGFR) transactivation, compared to vector control cells[79]. However, KP-10 did not increase cell migration, invasion and motility, or stimulate EGFR transactivaton in the ERα-positive T47D and MCF7 breast cancer cells[79]. Thus, in the presence of ERα, KP/KISS1R may function in an anti-metastatic capacity. In fact, Olbrich and others have shown that KP-10 inhibits bone-directed migration of KISS1R-positive MCF7 cells by down-regulating the expression of chemokine receptor CXCR4 and its ligand stomal derived factor (SDF)-1[80]. Thus, one can speculate that in normal healthy mammary epithelia, estrogen signaling through ERα is responsible for maintaining normal breast epithelial growth and function, hence maintaining KP/KISS1R signaling in check through transcriptional regulation of KISS1[81], and/or KISS1R (Figs. 2, 3). However, in certain cancers, where ERα expression is lost or silenced via DNA methylation as is the case in ERα-negative cancers[82, 83], the brake keeping KP/KISS1R signaling in check is removed. As a result, this disinhibition results in increased transcription of KISS1 and/or KISS1R, and consequently increases signaling through KISS1R leading to the induction of EMT, ultimately allowing for the mammary epithelial cells to acquire a more migratory and invasive phenotype[79] (Fig. 3). Overall, these studies have established that the ERα status of breast epithelia critically regulates the ability of KISS1R signaling to stimulate invasiveness.
Novel KP/KISS1R signaling pathways in breast cancer cells lacking the estrogen receptor, ERα. ERα has been found to negatively regulates KISS1R expression. KISS1R activation in these cells leads to epidermal growth factor (EGFR) transactivation via β-arrestin 2- and IQGAP1-mediated pathways. KISS1R activation can induce epithelial-mesenchymal transition (EMT), resulting in a decrease in E-cadherin expression and acquisition of a mesenchymal phenotype, characterized by actin cytoskeleton re-organization and stress fiber formation. Expression of mesenchymal markers (Snail/slug, N-cadherin, Vimentin) also increases. KP/KISS1R pathway can also activate RhoA. Blue solid lines represent KISS1R interacting proteins; black dashed lines represent EGFR interacting proteins.
Proposed model for ERα-dependent KP/KISS1R signaling in breast epithelia. In normal healthy mammary epithelia, estrogen signaling through ERα is responsible for maintaining normal breast epithelial growth and function, keeping KP/KISS1R signaling in check through transcriptional regulation of KISS and/or KISS1R. However, in breast cancer, when ERα expression is lost or silenced, this results in increased transcription of KISS1 and/or KISS1R, and increased receptor signaling and the induction of EMT, allowing epithelial cells to acquire a more migratory and invasive phenotype.
The mechanistic details by which ERα decreases KISS1R expression in breast cancer cells are currently unknown. A few studies have investigated how ERα regulates KISS1. In melanoma, activator protein 2 alpha (AP-2α) has been described as a possible positive transcriptional regulator of KISS1 in melanoma cells via interaction with the transcription factor, specificity 1 protein (SP1)[84]. However, in MDA-MB-231 breast cancer cells stably expressing ERα, E2 down-regulated the transcriptional activity of the KISS1 promoter independent of the ER element and SP1 by modulating the binding of the active RNA polymerase II at the KISS1 promotor[81]. Clearly, more studies are required to better understand how ERα exerts its regulatory action in breast cancer cells (ERα-positive versus negative), compared to its effects in some other cancers, where KISS1 functions as a metastasis-suppressor gene.
Molecular Mechanisms Regulating KISS1R Signaling in Breast Cancer
Currently, plasma KP levels in breast cancer patients are unknown. However, KP-10 immunoreactivity has been shown in aggressive MDA-MB-231 human breast cancer cells that are triple-negative (lacking ER, progesterone receptor and HER2), indicating that breast cancer cells can produce KPs. KISS1 mRNA expression in these cells has also been demonstrated[78]. The clinical data thus far suggests that KISS1 may be preferentially expressed in primary tumors, whereas KISS1R levels are higher in metastases[64, 78]. If this is the case, then the question arises as to how the receptor signals, independent of the ligand. One possibility is that KISS1R display constitutively activity[12, 85], and this has been previously described in breast cancer cells[86].
The mechanisms by which KP/KISS1R regulates breast cancer cell migration and invasion, two processes required for metastasis remain largely unknown. KP-10 can stimulate breast cancer cell migration and invasion of the ERα-negative MDA-MB-231 and Hs578T that endogenously express KISS1R, via transactivation of the EGFR[86]. EGFR is a well-known clinical target that is upregulated in numerous cancers, including breast cancer[87]. KP-10 stimulates EGFR transactivation and MMP-9 secretion and activity via a β-arrestin 2-dependent pathway [86]. In contrast, in HT-1080 human fibrosarcoma cells, where KP/KISS1R signaling is anti-invasive, KISS1 expression reduces MMP-9 production[88]. β-arrestins have emerged as important regulators of KISS1R signaling, cancer progression, invasion and metastasis[89, 90]. Currently we do not know if KISS1R activity regulates EGF secretion; nevertheless, EGFR can also be activated independently of its ligand[91]. Furthermore, KISS1R is able to directly associate with EGFR, and stimulation of breast cancer cells with either KP-10 or EGF can regulate the endocytosis of both receptors[86], providing evidence of receptor crosstalk. One can speculate that KISS1R binds to EGFR and promotes ligand-independent activation of EGFR, and this may positively regulate breast cancer cell invasiveness (Fig. 3).
Tumor cell extravasation is a key pre-requisite step during metastasis. Exogenous expression of KISS1R in ERα-negative SKBR3 cells stimulates the extravasation of tumor cells, using the chick chorioallantoic membrane assay, providing the first evidence that KISS1R signaling regulates breast cancer invasion in vivo[79]. KISS1R-mediated EGFR transactivation and migration of breast cancer cells was inhibited by the KISS1R antagonist, P-234, implicating a role for KISS1R activation in these processes[79]. The actin cytoskeletal scaffolding protein IQGAP1 has been identified as a KISS1R binding partner and is necessary for KISS1R-mediated transactivation of the EGFR in breast cancer cells[79]. IQGAP1 plays a role in tumorigenesis and is considered to be an oncogene[92-94]. IQGAP1 is over-expressed in trastuzumab-resistant human breast epithelial cells and contributes to resistance to anti-HER2 therapies[93]. IQGAP1 binds to a diverse array of signaling and structural proteins regulating multiple processes such as cell polarization, motility, invasion, maintenance of cytoskeletal architecture, and E-cadherin-mediated cell-cell adhesion[94]. Endogenous KISS1R is co-localized with endogenous IQGAP1 in lamellipodia of motile breast cancer cells and regulates breast cancer cell motility[79]. IQGAP1 also interacts with β-arrestin 2, and both proteins can facilitate the scaffolding of the MAPK signaling components[95]. Thus, the association of β-arrestin 2 and IQGAP1 may bring together individual signaling complexes within the same area of the cell to allow for the spatial regulation of multiple processes including cell migration.
Thus far, studies indicate that KISS1R signaling may correlate positively with breast tumor progression and metastatic potential[63, 78, 96]. However, it appears that KP-10 does not affect proliferation of breast epithelial or breast cancer cell lines expressing the receptor endogenously or exogenously[79, 97]. The effects of KISS1R signaling on non-malignant breast epithelia are not clear. KISS1R has been shown to be expressed in normal breast tissue[5], nevertheless, the role of KISS1R in the physiological development of mammary tissue is unknown. Recently, the effect of KP signaling in the immortilized, non-transformed mammary epithelial cell line MCF10A was examined. MCF10A cells form acinar structures in three-dimensional (3D) basement membrane cultures that retain characteristics of the glandular epithelium in vivo[98]. Interestingly, these cells are ERα-negative[99] and endogenously express KISS1R [63]. Treatment with KP-10 significantly stimulated MCF10A cells to form invasive structures compared to untreated cells in 3D Matrigel cultures[79]. Furthermore, stable expression of KISS1R in MCF10A cells significantly stimulated invasiveness independent of exogenous KP-10 and induced a mesenchymal phenotype[81]. Hence, it is possible that under pathological conditions such as breast cancer and upon loss of ERα, there is an upregulation of KP and/or KISS1R. This would stimulate the mammary epithelial cells to undergo EMT-like events, acquiring mesenchymal-like phenotypes and ultimately resulting in enhanced migration and invasion and thereby promoting metastasis (Fig. 2, 3)[100].
Lastly, two recent papers have shown there is an inverse relationship between the expression of KISS1 functioning as a metastasis suppressor and WASF3, an oncogene and a member of the Wiskott-Aldrich family of proteins[101, 102]. WASF3 promotes invasion and metastasis in breast cancer cells, which have undergone EMT[101-103]. Overexpression of WASF3 in non-invasive MCF7 and T47D human breast cancer cells, which typically do not exhibit an EMT-like phenotype, increases their invasiveness as a result of increased ZEB1/2 levels, which specifically suppresses the anti-invasion chromosome 1 miR-200a/200b/429 cluster[102]. This study showed that the upregulation of ZEB1/2 by WASF3 occurs due to a decreased expression of KISS1[102]. On the contrary, knockdown of WASF3 in breast cancer cells leads to reduced ZEB1 levels and increased miR-200 and E-cadherin levels, resulting in the loss of invasion potential with concomitant loss of matrix MMP-9 activity[101, 102]. Therefore, the loss of KISS1 which has been observed clinically in regional or distant breast cancer metastases[52,65,66] could result via WASF3-mediated ZEB1/2 upregulation, resulting in increased metastasis, implying that this signaling pathway may be a potential target to suppress invasion of breast cancer cells.
Conclusions
In this review article, we provide information as to the state of our current knowledge on the role of KP/ KISS1R signaling pathway in breast cancer. The complexity and tissue specificity of this signaling pathway remains a challenge for understanding and targeting this pathway. Emerging evidence clearly suggests that KISS1R plays critical roles in regulating signaling cascades that are involved in cell migration, invasion and metastasis, as in the case for breast cancer. The conflicting data for the role of the KISS1 and KISS1R in tumorigenesis could possibly be due the existence of an alternately spliced form(s) of these genes or possibly due to the epigenetically regulation of these genes. Currently, the methylation status of KISS1/KISS1R in breast cancer is unknown but this might potentially explain differences in expression patterns observed in the difference studies. A better understanding of the underlying mechanisms by which KISS1R signaling regulates cell invasiveness, cross-talks with other receptors such as growth factor receptors and the influence of the ERα status of breast epithelia on KISS1R signaling, could shed light on whether or not targeting KISS1R in breast cancer is a potentially useful therapy.
Abbreviations
GPCR: G protein-coupled receptor; KP: kisspeptin; KISS1R: kisspeptin receptor; EGFR: epidermal growth factor receptor; GRK: GPCR kinase; SP1: specificity 1 protein; EMT: epithelial-mesenchymal transition; MMP: matrix metalloproteinase; ER: estrogen receptor; E2: Estradiol.
Acknowledgements
The authors thank Dr. Jeff Dixon for valuable comments and suggestions. This work was conducted with funds to M.B. from the Canadian Institutes of Health Research (CIHR) grant MOP 107972 and funds awarded to A.V.B by the Natural Sciences and Engineering Research Council of Canada (NSERC) RGPIN/327334-2011. M.B. is a recipient of a CIHR New Investigator Salary Award. D.C. is a recipient of studentships from the Translational Breast Cancer Research Unit and the CIHR Strategic Training Program in Cancer Research and Technology Transfer, London Regional Cancer Program. SGD.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Beck BH, Welch DR. The KISS1 metastasis suppressor: a good night kiss for disseminated cancer cells. Eur J Cancer. 2005;46:1283-9
2. Lee JH, Welch DR. Identification of highly expressed genes in metastasis-suppressed chromosome 6/human malignant melanoma hybrid cells using subtractive hybridization and differential display. Int J Cancer. 1997;71:1035-44
3. Kotani M, Detheux M, Vandenbogaerde A, Communi D, Vanderwinden JM, Le Poul E. et al. The metastasis suppressor gene KiSS-1 encodes kisspeptins, the natural ligands of the orphan G protein-coupled receptor GPR54. The Journal of biological chemistry. 2001;276:34631-6
4. Mead EJ, Maguire JJ, Kuc RE, Davenport AP. Kisspeptins are novel potent vasoconstrictors in humans, with a discrete localization of their receptor, G protein-coupled receptor 54, to atherosclerosis-prone vessels. Endocrinology. 2007;148:140-7
5. Ohtaki T, Shintani Y, Honda S, Matsumoto H, Hori A, Kanehashi K. et al. Metastasis suppressor gene KiSS-1 encodes peptide ligand of a G-protein-coupled receptor. Nature. 2001;411:613-7
6. Gutierrez-Pascual E, Leprince J, Martinez-Fuentes AJ, Segalas-Milazzo I, Pineda R, Roa J. et al. In vivo and in vitro structure-activity relationships and structural conformation of Kisspeptin-10-related peptides. Mol Pharmacol. 2009;76:58-67
7. Lee JH, Miele ME, Hicks DJ, Phillips KK, Trent JM, Weissman BE. et al. KiSS-1, a novel human malignant melanoma metastasis-suppressor gene. J Natl Cancer Inst. 1996;88:1731-7
8. Muir AI, Chamberlain L, Elshourbagy NA, Michalovich D, Moore DJ, Calamari A. et al. AXOR12, a novel human G protein-coupled receptor, activated by the peptide KiSS-1. The Journal of biological chemistry. 2001;276:28969-75
9. Kirby HR, Maguire JJ, Colledge WH, Davenport AP. International Union of Basic and Clinical Pharmacology. LXXVII. Kisspeptin receptor nomenclature, distribution, and function. Pharmacol Rev. 2010;62:565-78
10. Clements MK, McDonald TP, Wang R, Xie G, O'Dowd BF, George SR. et al. FMRFamide-related neuropeptides are agonists of the orphan G-protein-coupled receptor GPR54. Biochem Biophys Res Commun. 2001;284:1189-93
11. Babwah AV, Pampillo M, Min L, Kaiser UB, Bhattacharya M. Single-cell analyses reveal that KISS1R-expressing cells undergo sustained kisspeptin-induced signaling that is dependent upon an influx of extracellular Ca2+. Endocrinology. 2012;153:5875-87 doi:10.1210/en.2012-1747
12. Pampillo M, Camuso N, Taylor JE, Szereszewski JM, Ahow MR, Zajac M. et al. Regulation of GPR54 signaling by GRK2 and {beta}-arrestin. Mol Endocrinol. 2009;23:2060-74
13. Szereszewski JM, Pampillo M, Ahow MR, Offermanns S, Bhattacharya M, Babwah AV. GPR54 regulates ERK1/2 activity and hypothalamic gene expression in a Galpha(q/11) and beta-arrestin-dependent manner. PLoS One. 2010;5:e12964
14. Castano JP, Martinez-Fuentes AJ, Gutierrez-Pascual E, Vaudry H, Tena-Sempere M, Malagon MM. Intracellular signaling pathways activated by kisspeptins through GPR54: do multiple signals underlie function diversity? Peptides. 2009;30:10-5 doi:10.1016/j.peptides.2008.07.025
15. Novaira HJ, Ng Y, Wolfe A, Radovick S. Kisspeptin increases GnRH mRNA expression and secretion in GnRH secreting neuronal cell lines. Molecular and cellular endocrinology. 2009;311:126-34 doi:10.1016/j.mce.2009.06.011
16. Freedman NJ, Lefkowitz RJ. Desensitization of G protein-coupled receptors. Recent Prog Horm Res. 1996;51:319-51 discussion 52-3
17. Krupnick JG, Benovic JL. The role of receptor kinases and arrestins in G protein-coupled receptor regulation. Annu Rev Pharmacol Toxicol. 1998;38:289-319
18. Bianco SD, Vandepas L, Correa-Medina M, Gereben B, Mukherjee A, Kuohung W. et al. KISS1R intracellular trafficking and degradation: effect of the Arg386Pro disease-associated mutation. Endocrinology. 2011;152:1616-26 doi:10.1210/en.2010-0903
19. Luttrell LM, Lefkowitz RJ. The role of beta-arrestins in the termination and transduction of G-protein-coupled receptor signals. J Cell Sci. 2002;115:455-65
20. Britten CD. PI3K and MEK inhibitor combinations: examining the evidence in selected tumor types. Cancer chemotherapy and pharmacology. 2013;71:1395-409 doi:10.1007/s00280-013-2121-1
21. Pinilla L, Aguilar E, Dieguez C, Millar RP, Tena-Sempere M. Kisspeptins and reproduction: physiological roles and regulatory mechanisms. Physiol Rev. 2012;92:1235-316
22. de Roux N, Genin E, Carel JC, Matsuda F, Chaussain JL, Milgrom E. Hypogonadotropic hypogonadism due to loss of function of the KiSS1-derived peptide receptor GPR54. Proc Natl Acad Sci U S A. 2003;100:10972-6
23. Seminara SB, Messager S, Chatzidaki EE, Thresher RR, Acierno JS Jr, Shagoury JK. et al. The GPR54 gene as a regulator of puberty. N Engl J Med. 2003;349:1614-27
24. Bilban M, Ghaffari-Tabrizi N, Hintermann E, Bauer S, Molzer S, Zoratti C. et al. Kisspeptin-10, a KiSS-1/metastin-derived decapeptide, is a physiological invasion inhibitor of primary human trophoblasts. J Cell Sci. 2004;117:1319-28
25. Yi T, Tan K, Cho SG, Wang Y, Luo J, Zhang W. et al. Regulation of embryonic kidney branching morphogenesis and glomerular development by KISS1 receptor (Gpr54) through NFAT2- and Sp1-mediated Bmp7 expression. The Journal of biological chemistry. 2010;285:17811-20
26. Goldberg SF, Miele ME, Hatta N, Takata M, Paquette-Straub C, Freedman LP. et al. Melanoma metastasis suppression by chromosome 6: evidence for a pathway regulated by CRSP3 and TXNIP. Cancer research. 2003;63:432-40
27. Navenot JM, Wang Z, Chopin M, Fujii N, Peiper SC. Kisspeptin-10-induced signaling of GPR54 negatively regulates chemotactic responses mediated by CXCR4: a potential mechanism for the metastasis suppressor activity of kisspeptins. Cancer research. 2005;65:10450-6
28. Shirasaki F, Takata M, Hatta N, Takehara K. Loss of expression of the metastasis suppressor gene KiSS1 during melanoma progression and its association with LOH of chromosome 6q16.3-q23. Cancer research. 2001;61:7422-5
29. Ringel MD, Hardy E, Bernet VJ, Burch HB, Schuppert F, Burman KD. et al. Metastin receptor is overexpressed in papillary thyroid cancer and activates MAP kinase in thyroid cancer cells. J Clin Endocrinol Metab. 2002;87:2399
30. Stathatos N, Bourdeau I, Espinosa AV, Saji M, Vasko VV, Burman KD. et al. KiSS-1/G protein-coupled receptor 54 metastasis suppressor pathway increases myocyte-enriched calcineurin interacting protein 1 expression and chronically inhibits calcineurin activity. J Clin Endocrinol Metab. 2005;90:5432-40
31. Gao GL, Liu LD, Zou XS, Chen WX. [Expression of KiSS-1, matrix metalloproteinase-9, nuclear factor-kappaBp65 in ovarian tumour]. Zhonghua Fu Chan Ke Za Zhi. 2007;42:34-8
32. Hata K, Dhar DK, Watanabe Y, Nakai H, Hoshiai H. Expression of metastin and a G-protein-coupled receptor (AXOR12) in epithelial ovarian cancer. Eur J Cancer. 2007;43:1452-9
33. Zhang C, Roepke TA, Kelly MJ, Ronnekleiv OK. Kisspeptin depolarizes gonadotropin-releasing hormone neurons through activation of TRPC-like cationic channels. J Neurosci. 2008;28:4423-34
34. Jiang T, Zhang SL, Lin B, Meng LR, Gao H. [Expression and clinical significance of KISS-1 and GPR54 mRNA in endometrial carcinoma]. Zhonghua Zhong Liu Za Zhi. 2005;27:229-31
35. Cebrian V, Fierro M, Orenes-Pinero E, Grau L, Moya P, Ecke T. et al. KISS1 methylation and expression as tumor stratification biomarkers and clinical outcome prognosticators for bladder cancer patients. The American journal of pathology. 2011;179:540-6 doi:10.1016/j.ajpath.2011.05.009
36. Sanchez-Carbayo M, Belbin TJ, Scotlandi K, Prystowsky M, Baldini N, Childs G. et al. Expression profiling of osteosarcoma cells transfected with MDR1 and NEO genes: regulation of cell adhesion, apoptosis, and tumor suppression-related genes. Lab Invest. 2003;83:507-17
37. Dhar DK, Naora H, Kubota H, Maruyama R, Yoshimura H, Tonomoto Y. et al. Downregulation of KiSS-1 expression is responsible for tumor invasion and worse prognosis in gastric carcinoma. Int J Cancer. 2004;111:868-72
38. Guan-Zhen Y, Ying C, Can-Rong N, Guo-Dong W, Jian-Xin Q, Jie-Jun W. Reduced protein expression of metastasis-related genes (nm23, KISS1, KAI1 and p53) in lymph node and liver metastases of gastric cancer. Int J Exp Pathol. 2007;88:175-83
39. Ikeguchi M, Yamaguchi K, Kaibara N. Clinical significance of the loss of KiSS-1 and orphan G-protein-coupled receptor (hOT7T175) gene expression in esophageal squamous cell carcinoma. Clin Cancer Res. 2004;10:1379-83
40. Masui T, Doi R, Mori T, Toyoda E, Koizumi M, Kami K. et al. Metastin and its variant forms suppress migration of pancreatic cancer cells. Biochem Biophys Res Commun. 2004;315:85-92
41. Zohrabian VM, Nandu H, Gulati N, Khitrov G, Zhao C, Mohan A. et al. Gene expression profiling of metastatic brain cancer. Oncol Rep. 2007;18:321-8
42. Martinez-Fuentes AJ, Molina M, Vazquez-Martinez R, Gahete MD, Jimenez-Reina L, Moreno-Fernandez J. et al. Expression of functional KISS1 and KISS1R system is altered in human pituitary adenomas: evidence for apoptotic action of kisspeptin-10. European journal of endocrinology / European Federation of Endocrine Societies. 2011;164:355-62 doi:10.1530/eje-10-0905
43. Gottsch ML, Clifton DK, Steiner RA. From KISS1 to kisspeptins: An historical perspective and suggested nomenclature. Peptides. 2009;30:4-9
44. Makri A, Pissimissis N, Lembessis P, Polychronakos C, Koutsilieris M. The kisspeptin (KiSS-1)/GPR54 system in cancer biology. Cancer Treat Rev. 2008;34:682-92
45. Cho SG, Li D, Tan K, Siwko SK, Liu M. KiSS1 and its G-protein-coupled receptor GPR54 in cancer development and metastasis. Cancer Metastasis Rev. 2012;31:585-91
46. Li N, Wang HX, Zhang J, Ye YP, He GY. KISS-1 inhibits the proliferation and invasion of gastric carcinoma cells. World journal of gastroenterology: WJG. 2012;18:1827-33 doi:10.3748/wjg.v18.i15.1827
47. Navenot JM, Fujii N, Peiper SC. Activation of Rho and Rho-associated kinase by GPR54 and KiSS1 metastasis suppressor gene product induces changes of cell morphology and contributes to apoptosis. Molecular pharmacology. 2009;75:1300-6 doi:10.1124/mol.109.055095
48. Cho SG, Yi Z, Pang X, Yi T, Wang Y, Luo J. et al. Kisspeptin-10, a KISS1-derived decapeptide, inhibits tumor angiogenesis by suppressing Sp1-mediated VEGF expression and FAK/Rho GTPase activation. Cancer research. 2009;69:7062-70 doi:10.1158/0008-5472.can-09-0476
49. Canbay E, Ergen A, Bugra D, Yamaner S, Eraltan IY, Buyukuncu Y. et al. Kisspeptin-54 Levels are increased in Patients with Colorectal Cancer. World J Surg. 2012;2012:3
50. Jarzabek K, Kozlowski L, Milewski R, Wolczynski S. KiSS1/GPR54 and estrogen-related gene expression profiles in primary breast cancer. Oncol Lett. 2012;3:930-4
51. Katagiri F, Nagai K, Kida A, Tomita K, Oishi S, Takeyama M. et al. Clinical significance of plasma metastin level in pancreatic cancer patients. Oncol Rep. 2009;21:815-9
52. Kostadima L, Pentheroudakis G, Pavlidis N. The missing kiss of life: transcriptional activity of the metastasis suppressor gene KiSS1 in early breast cancer. Anticancer Res. 2007;27:2499-504
53. Martins CM, Fernandes BF, Antecka E, Di Cesare S, Mansure JJ, Marshall JC. et al. Expression of the metastasis suppressor gene KISS1 in uveal melanoma. Eye (Lond). 2008;22:707-11
54. Moya P, Esteban S, Fernandez-Suarez A, Maestro M, Morente M, Sanchez-Carbayo M. KiSS-1 methylation and protein expression patterns contribute to diagnostic and prognostic assessments in tissue specimens for colorectal cancer. Tumour Biol. 2013;34:471-9
55. Nagai K, Doi R, Katagiri F, Ito T, Kida A, Koizumi M. et al. Prognostic value of metastin expression in human pancreatic cancer. J Exp Clin Cancer Res. 2009;28:9
56. Prentice LM, Klausen C, Kalloger S, Kobel M, McKinney S, Santos JL. et al. Kisspeptin and GPR54 immunoreactivity in a cohort of 518 patients defines favourable prognosis and clear cell subtype in ovarian carcinoma. BMC Med. 2007;5:33
57. Ikeguchi M, Hirooka Y, Kaibara N. Quantitative reverse transcriptase polymerase chain reaction analysis for KiSS-1 and orphan G-protein-coupled receptor (hOT7T175) gene expression in hepatocellular carcinoma. J Cancer Res Clin Oncol. 2003;129:531-5
58. Hou YK, Wang Y, Cong WM, Wu MC. [Expression of tumor metastasis-suppressor gene KiSS-1 and matrix metalloproteinase-9 in portal vein tumor thrombus of hepatocellular carcinoma]. Ai Zheng. 2007;26:591-5
59. Schmid K, Wang X, Haitel A, Sieghart W, Peck-Radosavljevic M, Bodingbauer M. et al. KiSS-1 overexpression as an independent prognostic marker in hepatocellular carcinoma: an immunohistochemical study. Virchows Arch. 2007;450:143-9
60. Lee JH, Welch DR. Suppression of metastasis in human breast carcinoma MDA-MB-435 cells after transfection with the metastasis suppressor gene, KiSS-1. Cancer research. 1997;57:2384-7
61. Ross DT, Scherf U, Eisen MB, Perou CM, Rees C, Spellman P. et al. Systematic variation in gene expression patterns in human cancer cell lines. Nat Genet. 2000;24:227-35
62. Chambers AF. MDA-MB-435 and M14 cell lines: identical but not M14 melanoma? Cancer research. 2009;69:5292-3
63. Cho SG, Wang Y, Rodriguez M, Tan K, Zhang W, Luo J. et al. Haploinsufficiency in the prometastasis Kiss1 receptor Gpr54 delays breast tumor initiation, progression, and lung metastasis. Cancer research. 2011;71:6535-46
64. Martin TA, Watkins G, Jiang WG. KiSS-1 expression in human breast cancer. Clin Exp Metastasis. 2005;22:503-11
65. Xie F, Yang H, Wang S, Zhou B, Tong F, Yang D. et al. A logistic regression model for predicting axillary lymph node metastases in early breast carcinoma patients. Sensors (Basel). 2012;12:9936-50
66. Mooez S, Malik FA, Kayani MA, Rashid R, Zahid A, Khan A. Expressional alterations and transcript isoforms of metastasis suppressor genes (KAI1 and KiSS1) in breast cancer patients. Asian Pac J Cancer Prev. 2011;12:2785-91
67. Ulasov IV, Kaverina NV, Pytel P, Thaci B, Liu F, Hurst DR. et al. Clinical significance of KISS1 protein expression for brain invasion and metastasis. Cancer. 2012;118:2096-105
68. Stark AM, Tongers K, Maass N, Mehdorn HM, Held-Feindt J. Reduced metastasis-suppressor gene mRNA-expression in breast cancer brain metastases. J Cancer Res Clin Oncol. 2005;131:191-8
69. Guttilla IK, Adams BD, White BA. ERalpha, microRNAs, and the epithelial-mesenchymal transition in breast cancer. Trends Endocrinol Metab. 2012;23:73-82
70. Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA. et al. Molecular portraits of human breast tumours. Nature. 2000;406:747-52
71. Stingl J. Estrogen and progesterone in normal mammary gland development and in cancer. Horm Cancer. 2011;2:85-90
72. Yan W, Cao QJ, Arenas RB, Bentley B, Shao R. GATA3 inhibits breast cancer metastasis through the reversal of epithelial-mesenchymal transition. The Journal of biological chemistry. 2010;285:14042-51
73. Rosen JM. On hormone action in the mammary gland. Cold Spring Harb Perspect Biol. 2012 4
74. Smith JT, Cunningham MJ, Rissman EF, Clifton DK, Steiner RA. Regulation of Kiss1 gene expression in the brain of the female mouse. Endocrinology. 2005;146:3686-92
75. Franceschini I, Lomet D, Cateau M, Delsol G, Tillet Y, Caraty A. Kisspeptin immunoreactive cells of the ovine preoptic area and arcuate nucleus co-express estrogen receptor alpha. Neurosci Lett. 2006;401:225-30
76. Clarke RB, Anderson E, Howell A. Steroid receptors in human breast cancer. Trends Endocrinol Metab. 2004;15:316-23
77. Parl FF, Schmidt BP, Dupont WD, Wagner RK. Prognostic significance of estrogen receptor status in breast cancer in relation to tumor stage, axillary node metastasis, and histopathologic grading. Cancer. 1984;54:2237-42
78. Marot D, Bieche I, Aumas C, Esselin S, Bouquet C, Vacher S. et al. High tumoral levels of Kiss1 and G-protein-coupled receptor 54 expression are correlated with poor prognosis of estrogen receptor-positive breast tumors. Endocr Relat Cancer. 2007;14:691-702
79. Cvetkovic D, Dragan M, Leith SJ, Mir ZM, Leong HS, Pampillo M. et al. KISS1R Induces Invasiveness of Estrogen Receptor-Negative Human Mammary Epithelial and Breast Cancer Cells. Endocrinology. 2013
80. Olbrich T, Ziegler E, Turk G, Schubert A, Emons G, Grundker C. Kisspeptin-10 inhibits bone-directed migration of GPR54-positive breast cancer cells: Evidence for a dose-window effect. Gynecol Oncol. 2010;119:571-8
81. Huijbregts L, de Roux N. KISS1 is down-regulated by 17beta-estradiol in MDA-MB-231 cells through a nonclassical mechanism and loss of ribonucleic acid polymerase II binding at the proximal promoter. Endocrinology. 2010;151:3764-72
82. Izadi P, Noruzinia M, Karimipoor M, Karbassian MH, Akbari MT. Promoter hypermethylation of estrogen receptor alpha gene is correlated to estrogen receptor negativity in Iranian patients with sporadic breast cancer. Cell J. 2012;14:102-9
83. Shi JF, Li XJ, Si XX, Li AD, Ding HJ, Han X. et al. ERalpha positively regulated DNMT1 expression by binding to the gene promoter region in human breast cancer MCF-7 cells. Biochem Biophys Res Commun. 2012;427:47-53
84. Mitchell DC, Stafford LJ, Li D, Bar-Eli M, Liu M. Transcriptional regulation of KiSS-1 gene expression in metastatic melanoma by specificity protein-1 and its coactivator DRIP-130. Oncogene. 2007;26:1739-47
85. Pampillo M, Babwah AV. Assessment of constitutive activity and internalization of GPR54 (KISS1-R). Methods Enzymol. 2010;484:75-93
86. Zajac M, Law J, Cvetkovic DD, Pampillo M, McColl L, Pape C. et al. GPR54 (KISS1R) transactivates EGFR to promote breast cancer cell invasiveness. PLoS One. 2011;6:e21599
87. Eccles SA. The epidermal growth factor receptor/Erb-B/HER family in normal and malignant breast biology. Int J Dev Biol. 2011;55:685-96
88. Yan C, Wang H, Boyd DD. KiSS-1 represses 92-kDa type IV collagenase expression by down-regulating NF-kappa B binding to the promoter as a consequence of Ikappa Balpha -induced block of p65/p50 nuclear translocation. The Journal of biological chemistry. 2001;276:1164-72
89. Li TT, Alemayehu M, Aziziyeh AI, Pape C, Pampillo M, Postovit LM. et al. Beta-arrestin/Ral signaling regulates lysophosphatidic acid-mediated migration and invasion of human breast tumor cells. Mol Cancer Res. 2009;7:1064-77
90. Fereshteh M, Ito T, Kovacs JJ, Zhao C, Kwon HY, Tornini V. et al. beta-Arrestin2 mediates the initiation and progression of myeloid leukemia. Proc Natl Acad Sci U S A. 2012;109:12532-7
91. Wang Y, Roche O, Xu C, Moriyama EH, Heir P, Chung J. et al. Hypoxia promotes ligand-independent EGF receptor signaling via hypoxia-inducible factor-mediated upregulation of caveolin-1. Proc Natl Acad Sci U S A. 2012;109:4892-7
92. Johnson M, Sharma M, Henderson BR. IQGAP1 regulation and roles in cancer. Cell Signal. 2009;21:1471-8
93. White CD, Li Z, Dillon DA, Sacks DB. IQGAP1 protein binds human epidermal growth factor receptor 2 (HER2) and modulates trastuzumab resistance. The Journal of biological chemistry. 2011;286:29734-47
94. Briggs MW, Sacks DB. IQGAP proteins are integral components of cytoskeletal regulation. EMBO Rep. 2003;4:571-4
95. Brown MD, Sacks DB. IQGAP1 in cellular signaling: bridging the GAP. Trends Cell Biol. 2006;16:242-9
96. Ulasov IV, Kaverina NV, Pytel P, Thaci B, Liu F, Hurst DR. et al. Clinical significance of KISS1 protein expression for brain invasion and metastasis. Cancer. 2002;118:2096-105
97. Ziegler E, Olbrich T, Emons G, Grundker C. Antiproliferative effects of kisspeptin10 depend on artificial GPR54 (KISS1R) expression levels. Oncol Rep. 2013;29:549-54
98. Debnath J, Mills KR, Collins NL, Reginato MJ, Muthuswamy SK, Brugge JS. The role of apoptosis in creating and maintaining luminal space within normal and oncogene-expressing mammary acini. Cell. 2002;111:29-40
99. Wang J, Gildea JJ, Yue W. Aromatase overexpression induces malignant changes in estrogen receptor alpha negative MCF-10A cells. Oncogene. 2012
100. Hugo H, Ackland ML, Blick T, Lawrence MG, Clements JA, Williams ED. et al. Epithelial--mesenchymal and mesenchymal--epithelial transitions in carcinoma progression. J Cell Physiol. 2007;213:374-83
101. Teng Y, Liu M, Cowell JK. Functional interrelationship between the WASF3 and KISS1 metastasis-associated genes in breast cancer cells. Int J Cancer. 2011;129:2825-35
102. Teng Y, Mei Y, Hawthorn L, Cowell JK. WASF3 regulates miR-200 inactivation by ZEB1 through suppression of KISS1 leading to increased invasiveness in breast cancer cells. Oncogene. 2013
103. Kulkarni S, Augoff K, Rivera L, McCue B, Khoury T, Groman A. et al. Increased expression levels of WAVE3 are associated with the progression and metastasis of triple negative breast cancer. PLoS One. 2012;7:e42895
Author contact
![]() Corresponding author: Dr. Moshmi Bhattacharya, Department of Physiology and Pharmacology, Medical Sciences Building, MSB 216, Schulich School of Medicine and Dentistry, University of Western Ontario, London, Ontario, Canada, N6A 5C1. Tel: 519-661-2111, ext. 82970; Fax: 519-661-3827 E-mail: moshmi.bhattacharyauwo.ca.
Corresponding author: Dr. Moshmi Bhattacharya, Department of Physiology and Pharmacology, Medical Sciences Building, MSB 216, Schulich School of Medicine and Dentistry, University of Western Ontario, London, Ontario, Canada, N6A 5C1. Tel: 519-661-2111, ext. 82970; Fax: 519-661-3827 E-mail: moshmi.bhattacharyauwo.ca.