Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2013; 4(8):606-613. doi:10.7150/jca.6453 This issue Cite
Research Paper
Normal Sequence and Activity but Reduced Levels of DNA-Pkcs in Human Lymphoblastic Cells Implicate Impaired Protein Stability with Radiosensitive Phenotype
Seow Fong Yap, Cynthia SK Boo, Susan LE Loong ![]() , Rajamanickam Baskar
, Rajamanickam Baskar ![]()
Department of Radiation Oncology, Division of Cellular and Molecular Research, National Cancer Centre, Singapore.
Received 2013-4-12; Accepted 2013-8-3; Published 2013-9-7
Abstract
Background: Non-homologous end joining (NHEJ) is the main repair pathway for DNA double strand breaks (DSBs) induced by ionizing radiation in mammalian cells. Subsets of cancer patients are hypersensitive to radiotherapy after standard doses. We sought to determine the radiosensitivity of human lymphoblastic cells (LB0005) for the abnormality in NHEJ components.
Methods: Lymphoblastic (LB0005) cells are derived from an adult cancer patient with late radionecrosis. A low magnesium in vitro DNA-end joining assay was performed to examine for any defect in NHEJ activity. Single-nucleotide polymorphism (SNP) and sequence analysis were performed to examine for abnormality if any, in the genetic sequence of known NHEJ components.
Results: LB0005 cells showed a gain of functional abnormality in the NHEJ pathway. While genetic sequence analysis showed no apparent mutational variations in the known classical NHEJ components, DNA-PKcs (DNA-dependent protein kinase catalytic subunit) protein is reduced in quantity compared to normal control, in spite of higher transcript levels.
Conclusions: Taken together cells derived from a radiosensitive patient showed an abnormality in NHEJ activity. Proteins other than the classical NHEJ factors may regulate the NHEJ activity. Furthermore, the defect in theses regulatory proteins may have an impact on the stability of DNA-PKcs.
Keywords: DNA-PKcs, Non-homologous end joining, NHEJ
Introduction
Radiotherapy (RT) continues to be an important therapeutic modality for the treatment of cancer [1, 2]. Radiation induced DNA damage response is a complex signaling process involving a variety of cellular events, which rapidly activate either cell cycle arrest to facilitate DNA repair or if the DNA damage is too extensive to be repaired, induce apoptosis [3, 4]. The critical target of radiation has been generally considered to be DNA, including base damage, single strand breaks (SSBs), double strand breaks (DSBs) of varying complexity and DNA cross links. DSBs are the most responsible factor involved in radiation induced cell death. However, the response mechanisms to maintain the genomic integrity by DNA repair depend on the extent of genotoxic damage and the cell type. Cells that are subjected to double strand breaks (DSBs) in their DNA activate cell cycle checkpoints and alter specific gene transcription patterns to allow the DNA repair machinery to correct the damage and preserve the integrity of the genome. Two major repair pathways, HR (homologous recombination) and NHEJ (non-homologous end joining) efficiently repair the most of DSBs [5]. Furthermore, in mammalian cells 80-90% of DSBs repair involves NHEJ [6, 7], a process that requires Ku proteins (Ku70 and K80), DNA-PKcs, Artemis, XRCC4, DNA ligase IV and XLF as hitherto identified core components [5]. Ku proteins are the first one to recognize and bind at the DSB's free DNA ends [8]. The hetero-dimeric Ku70/Ku80 protein, once bound to DNA ends (Ku: DNA), improves the affinity of the relevant nuclease (Artemis), polymerase μ and λ, and ligase (XRCC4, DNA ligase IV) for each DNA end on each strand [9, 10].
Disruption of the DNA damage response pathways in human cells leads to genomic instability and an increased risk of cancer progression [11, 12]. During the past few years, many researchers have studied how these damage signals coordinately execute cellular responses to DNA damage, but much less are known about the mechanisms that initiate early events of DNA damage then spread the damage signals throughout the cell. We have established five lymphoblastoid cell lines (LB0001-LB0005) derived from adult individuals who presented with cancer at <50 years of age and subsequently developed late radionecrosis after standard radiotherapy [13]. Two of these cell lines, LB0004 and LB0005 demonstrated increased in vitro radiation sensitivity [13]. More recently we have shown that cell extracts isolated from the LB0004 and LB0005 displayed defects in the in vitro end joining of noncohesive termini compared with normal cell extracts [14]. This defect was seen in the absence of deoxyribonucleoside triphosphates (dNTPs, substrates for the replication of DNA) and further sensitive to inhibition by wortmannin, an inhibitor of phosphoinositide 3-kinases related enzymes such as DNA-PK (DNA-dependent protein kinase). Sequencing of the joined products revealed that the process was error prone with a greater frequency of deletions compared with that observed in normal controls [14]. In the present study, we have observed that the lymphoblastic (LB0005) cells derived from an adult cancer patient with late radionecrosis after standard radiotherapy has a gain of functional abnormality in the NHEJ pathway.
Materials and Methods
Cell lines and culture
SNC3, control lymphoblastic cells with normal in vitro radiation sensitivity and DNA-DSB repair kinetics was derived by Epstein-Barr virus immortalization from a healthy volunteer [14]. As a negative control, we used M059J, a radiosensitive human glioma cells with no DNA-PKcs expression [15] and reduced NHEJ in vitro [16]. Cells were cultured in DMEM medium supplemented with 10% foetal bovine serum and and 1% of penicillin and streptomycin (GIBCO BRL, Carlsbad, CA, USA). LB0005 cells were derived from a cancer patient who developed late radionecrosis [13]. SNC3 and LB0005 cells were grown in suspension in medium consisting of RPMI 1640 with 10-20% foetal bovine serum, 2mM L-glutamine, and 1% of penicillin and streptomycin, 0.2mM Sodium pyruvate (GIBCO BRL, Carlsbad, CA, USA), 0.1mM Oxalo acetic acid and 6.25mM MOPS (Sigma, St Louis, MO, USA). The cultures were maintained at 37•C in a humidified 5% CO2 atmosphere.
Cell extracts preparation, Western blot analysis and Immunoprecipitation/ immunodepletion
Whole cell extract was prepared as described previously [14]. Briefly, cells were suspended in 2 volumes of hypotonic lysis buffer (10mM Tris.Cl, pH8.0, 1mM EDTA, 1mM DTT) and lysed by homogenization. After 20 min on ice, 0.5 volume of high salt buffer (50 mM Tris-HCl, pH 7.5, 1 M KCl, 2 mM EDTA, and 1 mM DTT) was added. Proteins were recovered by centrifugation at 42,000 rpm for 10 min. The supernatant was dialyzed against a glycerol-containing buffer (20 mM Tris-HCl, pH 8.0, 0.1 M KoAc, 20 % v/v glycerol, 0.5 mM EDTA, 1 mM DTT) for 2 h, then the cell extract was snap frozen and stored in aliquots at -80• C. For immunoprecipitation or immunodepletion analysis, whole cell extract (500 µg) was incubated with 2 µg of DNA-PKcs antibody or isotype control IgG2a (Dako, Glostrup, Denmark) in 20 mM HEPES-KOH (pH7.5), 0.5 mM MgCl2, 80 mM KCl, 1 mM DTT and 10% glycerol at 4•C for 2 h (depletion) or overnight (immunoprecipitation). The DNA-PKcs complex was subsequently removed using protein-A in agarose (Sigma, St Louis, MO, USA) and the remaining supernatant was used for NHEJ assay. Proteins removed were released from the protein-A agarose by adding SDS-sample buffer, boiled for 5 min and analyzed using SDS-PAGE. Antibodies against DNA-PKcs, Ku70, Ku80 (NeoMarkers, Fremont, CA, USA), XRCC4 (Acris Antibodies, Inc. San Diego, CA, USA), Ligase IV (AbD Serotec, Oxford, UK), XLF (Bethyl Laboratories, Inc. Montgomery, TX, USA), Artemis (Rockland Immunochemicals Inc. Gilbertsville, PA, USA), p53(DO-1) and phosphorylation at serine 15 of p53 (pS15-p53) (Santa Cruz Biotechnology, Santa Cruz, CA, USA), ATM and phosphorylation at serine 1981 (ATM-pS1981) (Cell Signaling Technology, Danvers, MA, USA) were analyzed by Western blot. For western blot, cell extract was separated by 8% SDS-PAGE, transferred to PVDF membrane and probed by ECL-Plus kit as recommended by the manufacturer (Amersham Biosciences, Piscataway, NJ, USA).
Non-homologous end joining assay
The NHEJ was assessed as described earlier [14]. Briefly, 20 µg of protein was mixed with 200 ng of pcDNA3 (EcoRV treated) in NHEJ buffer (20 mM HEPES-KOH (pH 7.5), 0.5 mM MgCl2, 80 mM KCl, 1 mM ATP, 1 mM DTT). Reaction was carried out at 25•C at different time points (0.5, 1, 2 and 4 h) and the reaction was stopped by adding 0.5% SDS, 0.05 mM EDTA and subsequently treated with 10 µg of proteinase-K at 37•C for 1h. The digested DNA was electrophoresed on 1.0% agarose gel, stained with SYBER green (Invitrogen Corporation , Carlsbad, CA, USA) and visualized using Syngene Gene snap UK gel doc system.
In vivo kinase assay (non-radioactive method)
Exponentially growing cells were exposed to (0Gy or 10Gy) of γ-rays using 137Cs source at a dose rate of 0.85 Gy/min (Gamma cell 40 Exactor, MDS Nordion, Ontario, Canada). The cells were then allowed to recover for 30 min at 37•C, harvested and washed with ice cold PBS. Cytoplasmic and nuclear cell fractions were isolated using NE-PER reagent as recommended by the manufacturer (Pierce Biotechnology, Rockford, IL, USA). The cytoplasmic and nuclear fractions were analyzed by Western blot using DNA-PKcs pS2056 (Abcam, Cambridge, MA, USA) antibody as described above.
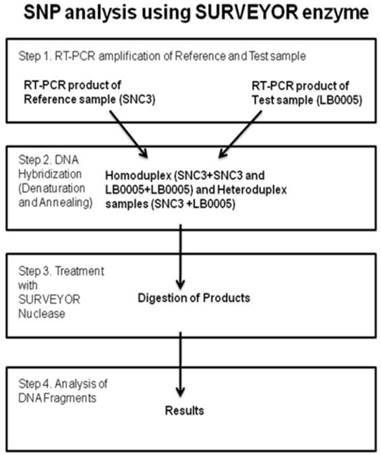
SURVEYOR analysis
Sequence analysis of the known classical NHEJ genes was monitored using SURVEYOR detection kit (Transgenomic, Inc. New Haven, CT, USA) (Figure 3). Briefly, primers were designed to RT-PCR overlapping DNA fragments spanning whole coding sequence of the gene of interest. SNC3 mRNA was used as normal control. Primer used were as follows, DNA-PKcs: (nucleotide position -23) 5'-gcgcgggagcgggac, (700) 5'-ttccatggacttagtgaagt, (600) 5'- tctgggtgaacttaagacc, (2500) 5'-aggttctttgtcttcttcag, (2388) 5'-ggatggatacctgaagact, (4300) 5'-gtcagggccatacaagttga, (3003) 5'-tgaaagtcaggatactgttg, (4900) 5'-ttcgagaggggaatctttg, (4200) 5'-tgaaagctctaaagatgtcc, (6100) 5'-ttgactcatttcctcactca, (5400) 5'-cgtgtatgaaatgttcagga, (6700) 5'-cgacaagggtctttataattt, (6580) 5'-tctttcatggacaggcttg, (8500) 5'-acgattgaagtcttgaagca, (7800) 5'-ccgatgtttgtggagacc, (9121)5'-gcggatcatgtaaggtaga, (8400) 5'-ggacccaataattgcaaaac, (9718) 5'-gcactgtctatcatcttcat, (9000) 5'-actgttacaaccaccttgc, (10878) 5'-taaacttccttctaaaggcc, (10770) 5'-tgatgtaagagctgaactag, (12410) 5'-gctttctatctgcagactc, Ku70: (-3) 5'-aacatgtcagggtgggagtc, (1189) 5'-aatgctgcaacctccttctcc, Ku80: (-25) 5'-accaaagcgcctgaggacc, (1494) 5'-aagctctgtgcagcagacac, (821) 5'-gacttggacagttgtggatgc, (2248) 5'-ccagcatcacagcgatggc, Artemis: (-32) 5'-tccaactatctccatagaccg, (1178) 5'-caaagagatagtcatcttcctcc, (990) 5'-tacctctgtcctgtgaacgc, (2128) 5'-gactgtcatctctgtgcagg, (490) 5'-tggataatacgttctgtgatcc, (765) 5'-atgcatggatctgagtgttgc, Ligase IV: (-27) 5'-taaacgagaagattcatcaccg, (1525) 5'-agttctttcatggtgcagcc, (1135) 5'-catgagactctgagaaagagg, (2779) 5'-tgagtctgccagatcagagg, (1499) 5'-tcatactctctctcgtgttgg, (1872) 5'-ttgtggttcatcatcaccacc, XRCC4: (-33) 5'-accggaagtagagtcacgg, (757) 5'-ttacttacagcagctgaagcc, (18) 5'-gaatccaccttgtttctgaacc, (1098) 5'-gctgctgacttgaaattctcc, (215) 5'-agcattgttgtcaggagcagg, (492) 5'-atgtgtgagtgctaaggaagc, (999) 5'-ctcatcaaagaggtcttctgg, XLF: (-32) 5'-gtgcggctaagagagtggg, (700) 5'-gtttgaggtatgaggatctcc, (170) 5'-agccaaggagctgaacaagc, (930) 5'-ccatcctcagcagctgagg. Equal amounts of SNC3 and LB0005 RT-PCR products using the identical pair of primers were mixed (LB0005-LB0005, LB0005-SNC3, SNC3-SNC3), denatured and renatured. Subsequently, the mixed DNA was treated with SURVEYOR nuclease. Mismatch site of the heteroduplex DNA was cleaved in both DNA strand using SURVEYOR endonuclease. The treated heteroduplex DNA was analyzed using 1.0% agarose gel. Performing similar reaction using primers spanning smaller region at the mismatch site subsequently narrowed any regions with mismatch down. Subsequently, these regions in LB0005 were sequenced and sequence variations identified.
Cartoon depicting the method of Single Nucleotide Polymorphism (SNP) analysis using the SURVEYOR enzyme.
Results
To establish LB0005 cells with NHEJ defect is gain or loss of function
In order to examine the NHEJ activity in DNA-PKcs-depleted LB0005 cells, we used an in vitro assay in low magnesium (0.5 mM) conditions which has been shown to be dependent on DNA ligase IV, and would thus test the integrity of DNA-PK dependent pathway of NHEJ (D-NHEJ) [17]. LB0005 cell extracts joined EcoRV digested with blunt-end pcDNA3 DNA more efficiently than the normal control cells (SNC3) (Figure 1A). Negative control cell, M059J with no expression of DNA-PKcs showed an inefficient rate of end-joining.
Immunodepletion of DNA-PKcs-complex reverses the fast DNA-joining activity of LB0005. (A) Time course study of NHEJ activity of SNC3, M059J and LB0005. (A) DNA-end joining activity of DNA-PKcs-depleted LB0005 cell extract. (B) Western blot analysis of the DNA-PKcs-depleted LB0005 cell extracts: DNA-PKcs and Ku70, Ku80 were pulled down by DNA-PKcs antibody. (s): supernatant and (b): beads. (C) NHEJ activity of DNA-PKcs-depleted LB0005 cell extract. DNA-joining activity of DNA-PKcs-depleted in LB0005 was lower than the control IgG2a-treated LB0005 cells. Lane1: SNC3, lane 2: DNA-PKcs-depleted LB0005 cell extract, lane 3: IgG2a treated LB0005 cell extract, lane 4: SNC3 cell extract was mixed with DNA-PKcs-depleted LB0005 cell extracts. (D) Quantitation of results from three independent experiments showing DNA-end joining activity of the DNA-PKcs-depleted LB0005 cell extract.
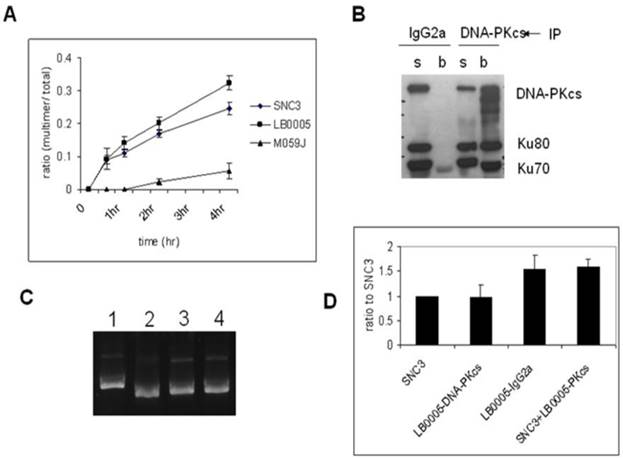
To further investigate if the NHEJ defect in LB0005 cells is a gain or loss of function abnormality, we set up another NHEJ assay by mixing the cell extracts of SNC3 and LB0005 cells. Since LB0005 cell extract has a high rate of DNA-end joining activity in vitro, NHEJ components were first depleted from LB0005 cell extracts using DNA-PKcs-specific antibody before mixing with SNC3 cell extracts. We chose DNA-PKcs as bait because DNA-PKcs is the core component of the NHEJ repair machinery. The large molecular size of DNA-PKcs (460 kDa) suggested it may not only serve as a kinase but could act as a scaffold protein providing a platform for the DSBs to be repaired. Furthermore, Western blot analysis showed LB0005 cells have reduced DNA-PKcs protein expression as compared to normal control SNC3 (Figure 2A). The mixing NHEJ assay showed that although depletion of DNA-PKcs was not complete (Figure 1B), a reduction in NHEJ activity was observed in the DNA-PKcs depleted LB0005 cell extract (Figure 1C and D). However, a control IgG2a treated LB0005 cell extract showed a higher NHEJ activity as compared to SNC3 cells, similar to untreated LB0005 cell extract. Furthermore, mixing the DNA-PKcs depleted LB0005 with SNC3 cell extracts in 1:1 showed an increased end-joining rate compared to the SNC3 cell extracts alone.
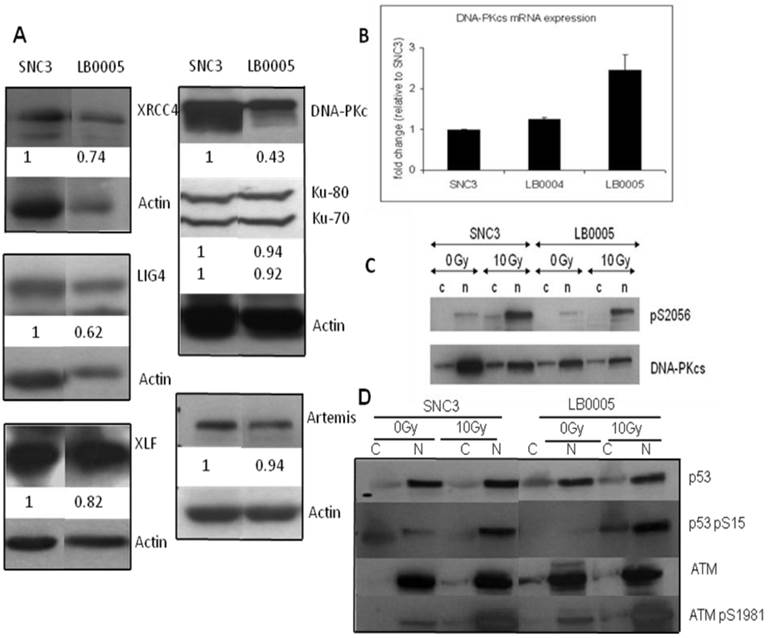
Expression of NHEJ core component in LB0005. (A) Western blot analysis showed expression of the NHEJ core component in LB0005. (B) Real time PCR analysis of DNA-PKcs expression in LB0005 cells. Real time PCR was carried out using TaqMan gene expression system. Expression of DNA-PKcs transcript in each cell line was first normalized against their GAPDH expression. The relative expression of DNA-PKcs was then expressed as a ratio to that of control cells (SNC3). (C) DNA-PKcs is phosphorylated upon γ-irradiation (0Gy or 10Gy). Phosphorylation of DNA-PKcs was detected using DNA-PKcs-pS2056 antibody by Western analysis. The blot is stripped and reprobed with DNA-PKcs antibody. (D) DNA damage response of LB0005 lymphoblastoid cells. Cytoplasmic (C) and nuclear (N) fractions.
Kinase activity of DNA-PKcs in vivo
Western blot analysis showed that LB0005 has reduced amount of DNA-PKcs protein expression as compared to SNC3 control. However, no genetic sequence variation was detected in PKcs between LB0005 when compared to SNC3. Thus, we next examined if the level of DNA-PKcs protein is correlated with its kinase activities by performing an in vivo non-radioactive kinase assay in LB00005 cells. Cells were first exposed to 0Gy or 10Gy of γ-radiation and allowed to recover for 1h at 37°C. Subsequently, cytoplasmic and nuclear cell extracts were isolated and analyzed using pS2056 and DNA-PKcs antibodies. S2056 has been chosen for the study because it has been shown as a site phosphorylated by DNA-PKcs, since lysate containing kinase dead DNA-PKcs does not phosphorylate S2056 even though its ATM was normal [19, 20]. The DNA-PKcs expressions in the LB0005 cells are reproducibly less than that of SNC3 (Figure 2C). However, after normalizing to that of total DNA-PKcs protein, the in vivo kinase activity of LB0005 cells did not show much difference than that of the control SNC3 cells.
Elucidating the genetic alteration in the known NHEJ components of LB0005 cells
Since the in vitro NHEJ assay suggested that LB0005 has a defect in the rate of end-joining in the classical DNA-PKcs-dependent NHEJ pathway, we therefore examined the protein expression and genetic sequence of the NHEJ components in LB0005 cells. Western blot analysis showed LB0005 cells expressed similar amounts of Ku70, Ku80, Artemis, XRCC4, Ligase IV and XLF of the expected molecular weight compared to SNC3 (Figure 2A). However, DNA-PKcs protein expression in LB0005 is lower when compared to that of SNC3, in spite of the fact that the DNA-PKcs transcript was expressed at a higher level (Figure 2B). Furthermore higher expression of DNA damage marker of phosphorylated form of p53 (p53-S15) was observed after 10Gy of γ-irradiation (Figure 2D).
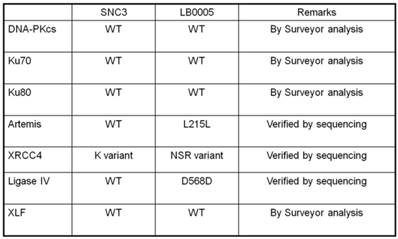
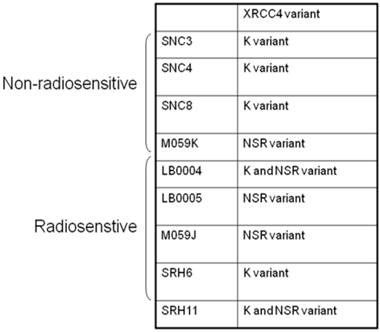
Abnormality of NHEJ components in LB0005 cells was also determined further by SNP and sequence analysis. Surveyor analysis was used to detect single nucleotide differences between LB0005 and SNC 3 cells, followed by partial sequencing (Table 1). SNC3 has normal NHEJ activity and was thus used as a control for the SNP and sequence analysis. Any identical SNPs or sequences to those obtained from SNC3 was considered normal (WT) and were not further analysed. There was no significant difference in the derived amino acid sequences of DNA-PKcs, Ku70, Ku80, Artemis, Ligase IV and XLF. For XRCC4, LB0005 differs from SNC3 sequence at bases 893-898. LB0005 cells showed an additional 6 nucleotides in the corresponding region, which influenced a change in the derived sequence of amino acids 298 to 300. The corresponding SNC3 sequence encodes for Lys (K variant), whereas the LB0005 sequence encodes for Asn-Ser-Arg (NSR variant) (Figure 3). In order to find out whether the two variants (K and NSR variants) had any relationship to the radiosensitivity phenotype, the corresponding region of XRCC4 sequence in different radiosensitive cells (LB0004, SRH6, SRH11 and M059J) and non-radiosensitive cells (SNC4, SNC8, M059K) were analyzed. LB0004, SRH6, SRH11, CNC4, SNC8 are lymphoblastoid cells and M059J and M059K are glioma cells. Although only the NSR variant was detected in the glioma cells, whereas both NSR and K variants were detected in the lymphoblastoid cells, suggesting the expression of XRCC4 variant may not be the cell-type specific. Furthermore, our results show that both radiosensitive and non-radiosensitive cells harbor sequence corresponding to either K or NSR variant or both indicating no correlation between radiosensitivity and the expression of XRCC4-K or NSR variant (Figure 4). This has also been independently confirmed by Mizuta et al [18], demonstrating that the core domain of XRCC4 encompassing amino acids 18-204, is sufficient to function in V(D)J recombination. Thus, amino acids 298-300 should be dispensable for the NHEJ activity of XRCC4.
Summary of NHEJ Genes of LB0005.
XRCC4 variants in selected radiosensitive and non-radiosensitive cell lines.
Discussion
Radiation therapy remains an important component of cancer treatment with approximately 50% of all cancer patients receiving radiation therapy during their course of illness; it contributes towards 40% of curative treatment for cancer [2]. The lethal target from ionizing radiation is the DNA-DSBs. For these NHEJ is the major repair pathway in mammalian cells. In our previous study, we have shown that in cell extracts derived from radiosensitive adult cancer patients showed an error-prone end-joining process [14]. In the present study, we have used an in vitro NHEJ assay in low magnesium conditions which has been shown to be dependent on DNA ligase IV and would thus test the integrity of DNA-PK dependent pathway of NHEJ [17]. We have shown that the cell extracts from radiosensitive LB0005 cells joined EcoRV digested blunt-ended pcDNA3-DNA more efficiently than the SNC3 (healthy volunteer control cells) and M059K, a DNA-PK proficient human glioma cells (data not shown) whereas, negative control cells M059J, deficient in DNA-PKcs, showed an inefficient rate of end-joining.
Results from NHEJ assay using DNA-PKcs-depleted LB0005 cell extracts suggested an abnormal function in DNA-PKcs complex or proteins that regulate the activity of DNA-PKcs complex. SNP analysis did not suggest any abnormality in the genetic sequence of LB0005, DNA-PKcs and other classical NHEJ components. Thus a mutation in a protein other than the NHEJ factors is involved in this process. The mixing of DNA-PKcs-depleted LB0005 and SNC3 cell extracts suggest a gain of function mutation in a regulatory protein for the DNA-PKcs complex. The mutation does not appear to impact on the kinase activity of LB0005-DNA-PKcs since in vivo kinase activity of DNA-PKcs is activated only after ionizing radiation treatment to similar extent of SNC3 (Figure 2C). The mutation does not interfere with the interaction of DNA-PKcs with Ku70, Ku80, XRCC4, Ligase IV and XLF, as suggested by immunoprecipitation using anti-DNA-Pkcs antibody (data not shown). However, Western blot analysis showed a less DNA-PKcs protein is expressed in LB0005 cells as compared to SNC3 cells. Intriguingly LB0005 cells expressed more DNA-PKcs mRNA than the SNC3 control cells. Therefore, our results suggest a defect that leads to instability of DNA-PKcs protein. Furthermore, the high mRNA and low protein expression of DNA-PKcs suggest that a proteosome-dependent degradation mechanism maybe involved. However, degradation of DNA-PKcs alone may not be the primary cause for NHEJ defects observed in LB0005 cells since a mere reduction in DNA-PKcs protein should cause an inefficient rate of DNA-end joining as suggested by the result from DNA-PKcs depleted extracts study. Therefore, it is likely this effect is secondary, that is, a mutation in a regulatory protein leads to abnormal function of DNA-PKcs and also to the degradation of DNA-PKcs. In support of this, it has been shown that overexpression of Bcr-Abl leads to degradation of DNA-PKcs in hematopoietic cells and CD34+ cells from patients with chronic myelogenous leukemia (CML) [21]. It was further shown that DNA-PKcs was down regulated by a proteasome-dependent degradation that requires the tyrosine kinase activity of Bcr-Abl [21].
Conclusions
In summary, lymphoblastic cells derived from a radiosensitive cancer patient showed a gain of function defect in DNA-end joining in vitro. From the present study, we postulate that the factors other than the classical NHEJ are important for the regulation of NHEJ activity in these lymphoblastic cells. Furthermore, the defect in this regulatory protein may have an impact on the stability of DNA-PKcs protein.
Acknowledgements
We thank Ms. Haslinda Kamis for technical assistance. This work was supported by grant from the National Medical Research Council (NMRC), Singapore (NMRC/1131/2007) to SL and RB is supported by the NMRC, Singapore (BNIG11nov004).
Authors' contributions
SFY, CB and RB participated in the design of experiments. SFY, CB and RB carried out the experiments. SFY, CB, RB and SL analyzed the data. RB and SL drafted the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
References
1. Baskar R, Yap SP, Chua KL, Itahana K. The diverse and complex roles of radiation on cancer treatment: therapeutic target and genome maintenance. Am J Cancer Res. 2012;2:372-382
2. Baskar R, Lee KA, Yeo R, Yeoh KW. Cancer and radiation therapy: current advances and future directions. Int J Med Sci. 2012;9:193-139
3. Bartek J, Lukas J. DNA damage checkpoints: from initiation to recovery or adaptation. Curr Opin Cell Biol. 2005;19:238-245
4. Harper JW, Elledge SJ. The DNA damage response: ten years after. Mol Cell. 2007;28:739-745
5. Collis SJ, DeWeese TL, Jeggo PA, Parker AR. The life and death of DNA-PK. Oncogene. 2005;24:949-961
6. Riballo E, Kuhne M, Rief N. et al. Pathway of double-strand break rejoining dependent upon ATM, Artemis, and proteins locating to gamma- H2AX foci. Mol. Cell. 2004;16:715-724
7. Kuhne M, Riballo E, Rief N. et al. Double-strand break repair defect in ATM-deficient cells contributes to radiosensitivity. Cancer Res. 2004;64:500-508
8. Ramsden DA, Gellert M. Ku protein stimulates DNA end joining by mammalian DNA ligases: a direct role for Ku in repair of DNA double-strand breaks. EMBO J. 1998;17:609-614
9. Chen L, Trujillo K, Sung P, Tomkinson AE. Interactions of the DNA ligase IV-XRCC4 complex with DNA ends and the DNA-dependent protein kinase. J Biol Chem. 2000;275:26196-26205
10. NickMcElhinny SA, Snowden CM, McCarville J, Ramsden DA. Ku recruits the XRCC4-ligase IV complex to DNA ends. Mol Cell Biol. 2000;20:2996-3003
11. Hoeijmakers JHJ. DNA damage, aging, and cancer. N Eng J Med. 2009;361:1475-1485
12. Stratton MR, Campbell PJ, Futreal PA. The cancer genome. Nature. 2009;458:719-724
13. Loong SL, Korzh S, Price A. Reduced DNA-dependent protein kinase activity in two cell lines derived from adult cancer patients with late radionecrosis. Oncogene. 2004;23:5562-5566
14. Tan WM, Paterson MC, Koo GC. et al. Increased but error-prone nonhomologous end joining in immortalized lymphoblastoid cell extracts from adult cancer patients with late radionecrosis. Int J Radiat Oncol Biol Phys. 2008;72:178-185
15. Lees-Miller SP, Godbout R, Chan DW. et al. Absence of p350 subunit of DNA-activated protein kinase from a radiosensitive human cell line. Science. 1995;267:1183-1185
16. Dai Y, Kysela B, Hanakahi LA. et al. Nonhomologous end joining and V(D)J recombination require an additional factor. Proc Natl Acad Sci USA. 2003;100:2462-2467
17. Wang H, Zeng ZC, Perrault AR. et al. Genetic evidence for the involvement of DNA ligase IV in the DNA-PK-dependent pathway of non-homologous end joining in mammalian cells. Nucleic Acids Res. 2001;29:1653-1660
18. Mizuta R, Cheng HL, Gao Y, Alt FW. Molecular genetic characterization of XRCC4 function. Int Immunol. 1997;9:1607-1613
19. Chen BP, Chan DW, Kobayashi J. et al. Cell Cycle Dependence of DNA-dependent Kinase phosphorylation in response to DNA double strand breaks. J Biol Chem. 2005;280:14709-14715
20. Oksenych V, Kumar V, Liu X. et al. Alt FW: Functional redundancy between the XLF and DNA-PKcs DNA repair factors in V(D) J recombination and non-homologous DNA end joining. Proc Natl Acad Sci USA. 2013;110:2234-2239
21. Deutsch E, Dugray A, AbdulKarim B. et al. BCR-ABL down-regulates the DNA repair protein DNA-PKcs. Blood. 2001;97:2084-2090
Author contact
![]() Corresponding author: R. Baskar M.Phil., Ph.D. Molecular Radiobiology Laboratory, Department of Radiation Oncology, Division of Cellular and Molecular Research, National Cancer Centre, 11- Hospital Drive, Singapore-169610, Singapore. Tel: +65- 6436 8315; Fax: +65-6222 8675. E-mail: r.baskarcom.sg OR Susan Loong, M.D., F.R.C.R., F.R.C.P. Department of Radiation Oncology, Division of Cellular and Molecular Research, National Cancer Centre, 11 Hospital Drive, Singapore 169610, Singapore. Tel: +65- 6321-4204; Fax: +65- 6222-8675. E-mail: drsloongcom.
Corresponding author: R. Baskar M.Phil., Ph.D. Molecular Radiobiology Laboratory, Department of Radiation Oncology, Division of Cellular and Molecular Research, National Cancer Centre, 11- Hospital Drive, Singapore-169610, Singapore. Tel: +65- 6436 8315; Fax: +65-6222 8675. E-mail: r.baskarcom.sg OR Susan Loong, M.D., F.R.C.R., F.R.C.P. Department of Radiation Oncology, Division of Cellular and Molecular Research, National Cancer Centre, 11 Hospital Drive, Singapore 169610, Singapore. Tel: +65- 6321-4204; Fax: +65- 6222-8675. E-mail: drsloongcom.