Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2013; 4(7):585-596. doi:10.7150/jca.7030 This issue Cite
Research Paper
The Autophagy Inhibitor Verteporfin Moderately Enhances the Antitumor Activity of Gemcitabine in a Pancreatic Ductal Adenocarcinoma Model
Elizabeth Donohue1, Anitha Thomas2, Norbert Maurer2, Irina Manisali2, Magali Zeisser-Labouebe2, Natalia Zisman2, Hilary J. Anderson1, Sylvia S. W. Ng3, Murray Webb2, Marcel Bally2,4, Michel Roberge1,2 ![]()
1. Department of Biochemistry and Molecular Biology, University of British Columbia;
2. Centre for Drug Research and Development, University of British Columbia;
3. Department of Pharmaceutical Sciences, BC Cancer Agency;
4. Department of Experimental Therapeutics, BC Cancer Agency, Vancouver, British Columbia, Canada
Received 2013-6-27; Accepted 2013-8-17; Published 2013-8-28
Abstract
Pancreatic ductal adenocarcinoma (PDAC) is highly resistant to chemotherapy. It has been described as requiring elevated autophagy for growth and inhibiting autophagy has been proposed as a treatment strategy. To date, all preclinical reports and clinical trials investigating pharmacological inhibition of autophagy have used chloroquine or hydroxychloroquine, which interfere with lysosomal function and block autophagy at a late stage. Verteporfin is a newly discovered autophagy inhibitor that blocks autophagy at an early stage by inhibiting autophagosome formation. Here we report that PDAC cell lines show variable sensitivity to verteporfin in vitro, suggesting cell-line specific autophagy dependence. Using image-based and molecular analyses, we show that verteporfin inhibits autophagy stimulated by gemcitabine, the current standard treatment for PDAC. Pharmacokinetic and efficacy studies in a BxPC-3 xenograft mouse model demonstrated that verteporfin accumulated in tumors at autophagy-inhibiting levels and inhibited autophagy in vivo, but did not reduce tumor volume or increase survival as a single agent. In combination with gemcitabine verteporfin moderately reduced tumor growth and enhanced survival compared to gemcitabine alone. While our results do not uphold the premise that autophagy inhibition might be widely effective against PDAC as a single-modality treatment, they do support autophagy inhibition as an approach to sensitize PDAC to gemcitabine.
Keywords: autophagy, pancreatic cancer, verteporfin, gemcitabine, chemosensitization
Introduction
Autophagy is a cellular degradation process that facilitates cytoplasmic turnover and maintains metabolic homeostasis in which double-membraned vesicles, termed autophagosomes, form around cytoplasmic components and are digested upon fusion with lysosomes. The degradation products are released to the cytoplasm for anabolism or energy production. Autophagy is upregulated in response to stresses such as starvation, hypoxia, and accumulation of damaged organelles or misfolded proteins (1,2) and can thus promote the survival of tumor cells in poorly vascularized and hypoxic tumors (3) or following cytotoxic treatments (4-6). Many preclinical studies have demonstrated that genetic or pharmacological inhibition of autophagy can enhance drug- and radiation-induced cytotoxicity in cell culture and in vivo (6-9).
Pancreatic ductal adenocarcinoma (PDAC) has a 5-year survival rate of only 6% and is notoriously resistant to chemotherapy (10). Elevated autophagy has been implicated in pancreatic malignancies (11-13). Yang et al (13) showed that some PDAC cell lines are inherently dependent on autophagy for growth, even in nutrient-rich conditions, and that inhibition of autophagic flux using chloroquine (CQ) resulted in moderately reduced growth of PDAC tumor xenografts in mice. The standard of care for patients with unresectable pancreatic cancer is gemcitabine, a nucleoside analog that provides only a modest survival benefit of 5 weeks (14,15), due largely to innate and acquired chemoresistance (16). Gemcitabine stimulates autophagy but whether the autophagic response protects tumor cells remains unclear (17-19). Two phase I/II clinical trials are recruiting patients for combinatorial treatment with gemcitabine and the CQ derivative hydroxychloroquine (HCQ), highlighting current interest in testing the role of autophagy in PDAC (Trial identifiers: NCT01506973 and NCT01128296).
CQ and HCQ inhibit autophagy non-selectively by neutralizing the pH of acidic intracellular vesicles, including lysosomes, thereby inhibiting lysosomal hydrolases responsible for autophagic degradation and causing the accumulation of undigested autophagosomes (20). A major issue for clinical trials is that the high micromolar concentrations of HCQ required to inhibit autophagy in vitro are inconsistently achieved in humans (21-23). Another is that prolonged CQ treatment can stimulate autophagosome biogenesis (24,25). Therefore, there is a need for other autophagy inhibitors. Verteporfin is an FDA-approved drug that was identified in a screen for chemicals that prevent autophagosome formation (26). Verteporfin is used clinically in photodynamic therapy, but it inhibits autophagy without light activation. Unlike CQ and HCQ, verteporfin inhibits autophagy at an early stage and does not cause autophagosome accumulation, and it inhibits autophagy in vitro with a lower IC50 of 1µM (26). In this study, we demonstrate that some PDAC cell lines are sensitive to verteporfin in vitro and that verteporfin moderately enhances the antitumor activity of gemcitabine in a BxPC-3 tumor xenograft model.
Methods and Materials
Reagents
Reagents were purchased as follows: cell culture reagents from Sigma-Aldrich unless stated otherwise; general laboratory chemicals from Sigma-Aldrich, Fisher Scientific, and BDH Inc.; verteporfin from Prestwick Chemical or from the United States Pharmacopeial Convention, Inc.; gemcitabine from Eli Lilly Canada Inc.; chloroquine from Sigma-Aldrich; 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (DSPE-mPEG2000) from Genzyme Pharmaceuticals.
Cell culture procedures
MCF-7 cells stably transfected with pEGFP-LC3 (27), BxPC-3, and SU86.86 cells were maintained in RPMI 1640 supplemented with 1mM Hepes. EGFP-LC3-expressing cells were supplemented with 400μg/ml G418. Capan 2, HS766T, Panc-1 cells and MIA PaCa-2 cells (American Type Culture Collection (ATTC)) were maintained in DMEM. Capan-1 and CFPAC-1 cells were maintained in IMDM (Gibco, #12440-053). All media were supplemented with 10% (v/v) fetal bovine serum (FBS), 100units/ml penicillin and 100µg/mL streptomycin (Gibco), and all cells were maintained at 37°C in 5% CO2.
BxPC-3 and SU86.86 cells used for the animal studies were purchased from ATCC and tested negative for mycoplasma contamination. Cells were grown in RPMI 1640 supplemented with 2mM L-glutamine and 10% FBS without antibiotics. Cells were sub-cultured once a week and cell cultures with passage number 3-10 were used for in vivo studies.
Cell viability and proliferation assays
Cells were seeded at 500-1000 cells per well in 96-well PerkinElmer View plates and grown overnight. The following day (t=0), four plates were identically treated as indicated for 1-7 days. At t = 1, 3, 5, and 7d, the appropriate plates were fixed and stained with 3% (v/v) paraformaldehyde in PBS containing 500ng/ml Hoechst 33342 for 15min at room temperature. Cells were identified and quantified using the Target Activation Bioapplication of a Cellomics ArrayScan VTI automated fluorescence imager. Fifteen fields were analyzed per well. Capan 1 and Capan 2 cells were seeded in 96-well Falcon tissue culture plates, and cell viability was measured by MTT assay (28).
Automated assay for monitoring autophagosome accumulation
Punctate EGFP-LC3 was monitored and quantified in MCF-7 EGFP-LC3 cells as described previously (26,27).
SDS-PAGE and immunoblotting
Tumor tissue was washed in PBS and chopped finely with a scalpel on ice, homogenized in a 10X volume of cold lysis buffer containing 20mM Tris-HCl, pH 7.5, 150mM NaCl, 1% (v/v) Triton X-100, 1mM EDTA, 1mM EGTA, 2.5mM sodium pyrophosphate, 1mM β-glycerophosphate, 1mM sodium orthovanadate, and 1x complete protease inhibitor cocktail (Roche). Lysates were centrifuged at 18,000 x g at 4°C, and supernatants were collected and quantified by Bradford assay (Bio-Rad). Lysates were normalized for protein content in SDS-PAGE sample buffer and analyzed by immunoblotting as in (26,27), using mouse α-GFP (1:7000, Roche #11814460001), mouse α-SQSTM1/p62 (1:250, Santa Cruz, sc-28359), mouse α-LC3 (1:1000, NanoTools 0260), rabbit α-β-tubulin (1:20000, Santa Cruz, sc-9104), HRP-conjugated goat α-mouse IgG (ThermoScientific 31430) and HRP-conjugated goat α-rabbit IgG (KPL 074-1506) antibodies.
Formulation of verteporfin for animal studies
Verteporfin was formulated in DSPE-mPEG2000 micelles. Briefly, 200mg verteporfin dissolved in 2ml DMSO was slowly added, with stirring, to 1500mg DSPE-mPEG2000 dissolved in 50ml PBS at pH 7.4. Stirring was continued for 1h at 23°C followed by dialysis overnight against PBS using Spectra/Por dialysis membranes (15,000 MWCO, Spectrum Laboratories Inc). The concentration of verteporfin was measured in triplicate and quantified against an external standard curve using a Waters® ACQUITY® UPLC equipped with a PDA detector. Separations were done using a C18 column (Waters® BEH; column size 50 x 2.1mm, particle size 1.7µm) and a mobile phase of 0.1% formic acid in water (A) and 0.1% formic acid in acetonitrile:methanol (1:1; B) [10-80% B over 2min at 0.5ml/min flow rate]. The concentration of verteporfin was adjusted to 2.25mg/ml with PBS followed by filter-sterilization and sterile vialing of the formulation. The concentration was reconfirmed before proceeding with the animal studies. The formulation was chemically and physically stable for an observation period of 4 weeks, which covered the duration of the animals studies as verified by UPLC and polarized light microscopy.
Animal husbandry
All animal studies were approved by the University of British Columbia Animal Care Committee and were performed in accordance with the Canadian Council on Animal Care guidelines (protocol #A10-0171). Animals were euthanized by CO2 asphyxiation when the tumor reached a weight of 800mg, an approved humane end point. Macroscopic necropsy was performed to assess signs of toxicity.
Verteporfin efficacy studies
Female Rag2M mice (20-25g) were inoculated subcutaneously in the centre of the lower back with 5 x 106 BxPC-3 or SU86.86 cells (1:1 RPMI:matrigel; 100µL volume; expressed as day 0). Tumors appeared within three weeks of implantation. Once the tumors were palpable, tumor growth was monitored by measuring tumor dimensions with digital calipers. When tumors reached 100-150mg (calculated according to the equation (length X width2)/2 converted to tumor weight in mg for each 1mm3), mice were randomized in groups of eight animals and treatment was initiated.
Verteporfin was administered i.p. Monday, Wednesday, Friday for 4 weeks at a dose of 45mg/kg (injection volume 400µl/20g mouse). Gemcitabine was administered i.p. once weekly (Monday) for 4 weeks at 120mg/kg or 240mg/kg (injection volume 200µl/20g mouse). Groups treated with both verteporfin and gemcitabine received gemcitabine 6h after verteporfin administration. This time was selected because maximum verteporfin tumor levels were achieved 8h post-administration and maximum gemcitabine tumor levels were observed 2h post-administration. Animals in the control group were treated with the delivery vehicle DSPE-mPEG2000 at the same concentration and schedule as verteporfin. Care was taken to house animals treated with verteporfin in dark conditions until the morning after treatment because verteporfin is a photosensitizer and exposure to bright light could be harmful. A One-Way ANOVA with Tukey's multiple comparison test was used to compare differences in tumor growth (GraphPad Prism Version 6.00).
Pharmacokinetic studies of verteporfin DSPE-mPEG2000 micelles in BxPC-3 tumor-bearing mice
Rag2M mice (20-25g; n=3) were inoculated subcutaneously with 5 x 106 BxPC-3 cells. Mice were injected i.p. with verteporfin at 45mg/kg when tumors were approximately 200-250mg. Mice were euthanized by CO2 inhalation, and blood and tumors were collected at 2, 8, 16 and 24h post administration of verteporfin. Plasma was prepared by centrifuging samples at 1000 x g for 15min at 4°C. Tumors were excised, rinsed in PBS, and snap-frozen in cryovials in liquid nitrogen and stored at -80°C. Tumors were sectioned while frozen and one half was used for determining verteporfin concentration by UPLC-MS/MS and the other for immunoblot analysis of p62 and LC3.
Plasma samples were thawed, homogenized and extracted with acetonitrile containing 0.1% formic acid. Protein precipitation and filtration was carried out using ISOLUTE® PPT+ protein precipitation plates (Biotage). Samples were analyzed using a Waters® ACQUITY® UPLC system with mass spectrometry detection. Separations were performed using an isocratic method where mobile phase A was 0.1% formic acid in water and B was 0.1% formic acid in acetonitrile (70% B for 2.5min followed by 95% B for the wash). Verteporfin regioisomer A (monoacid A form) was eluted at 1.77min and regioisomer B (monoacid B form) was eluted at 2.16min with a total run time of 4.5min per sample. The MS/MS system was operated with an ESI interface in a positive ionization mode. Quantification was performed using multiple reactions monitoring (MRM) mode with a precursor mass m/z of 719.27 and product mass m/z of 645.36. The levels of verteporfin were measured against external calibration standards prepared using the same process.
RESULTS
In vitro effects of verteporfin on PDAC cell lines
A panel of 8 human PDAC cell lines was exposed to 0-10µM verteporfin for up to 7 days and live cells were quantified using automated fluorescence microscopy to count nuclei or by measuring absorbance in an MTT assay. Four lines (Capan 1, Capan 2, HS766T, CFPAC-1) were insensitive to verteporfin at all concentrations (Fig. 1A). Panc-1 and MIA PaCa-2 cells grew normally in ≤ 5µM verteporfin, but cell proliferation was significantly inhibited in 10µM verteporfin. Notably, proliferation of BxPC-3 and SU86.86 cells was completely inhibited in 10µM verteporfin, and reduced by >50% in 5µM verteporfin (Fig. 1A).
Effect of long term verteporfin treatment on PDAC cell line viability in vitro. (A) The eight cell lines were treated with 0-10µM verteporfin (VP) for up to 7 days and cell proliferation was monitored at the indicated time points. Media and drugs were replenished every third day. Capan 1 and Capan 2 cell viability and proliferation was assayed using MTT and measuring OD 570. Other cell lines were stained with Hoechst 33342 and nuclei were quantified using a Cellomics ArrayScan VTI automated fluorescence microscope. (mean ± S.D. (error bars), n=4) (B) BxPC-3, SU86.86, and MIA PaCa-2 cells were exposed to vehicle control or 100nM bafilomycin A1 in complete medium for 4h. Lysates were collected and immunoblotted for LC3 or p62. β-tubulin was monitored as a loading control.
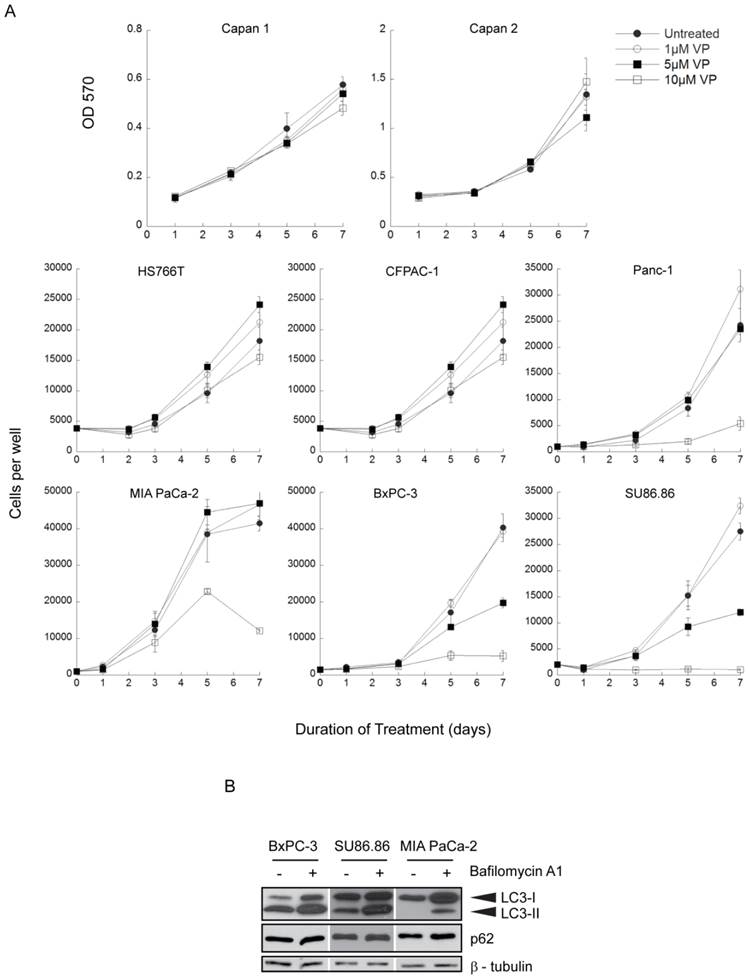
Activating K-Ras mutations are a hallmark of pancreatic cancer (29,30) and recent studies suggest Ras activation is a key to autophagy addiction (31,32). However, no correlation between K-Ras mutation and verteporfin sensitivity was observed in these cell lines: highly sensitive BxPC-3 and SU86.86 cells express wild type K-Ras and activated K-Ras, respectively (33-35) and Panc-1 cells with the same activated K-Ras mutation as SU86.86 cells are relatively insensitive to verteporfin (35). Other frequent PDAC mutations in the tumor suppressor genes p53 and DPC4 (29,34) also showed no noticeable correlation with verteporfin sensitivity.
Basal autophagy was monitored in highly verteporfin-sensitive BxPC-3 and SU86.86 cells and in relatively insensitive MIA PaCa-2 cells using two autophagy markers, LC3 and p62. During autophagy, the cytoplasmic protein LC3-I is lipidated to form LC3-II, which stably associates with the autophagosome membrane (36); inhibiting lysosomal degradation with bafilomycin A1 leads to increased LC3-I and LC3-II levels due to the accumulation of undigested autophagosomes. p62 is a protein degraded by autophagy and commonly monitored as a marker of autophagic flux (37). In the presence of bafilomycin A1 over a 4h period, LC3-I and LC3-II accumulated in all three cell lines compared to the respective control, but the relative accumulation between the three cell lines did not differ greatly (Fig. 1B). Additionally, p62 levels were unchanged in all three cell lines, indicating the autophagic flux over 4h was quite low (Fig. 1B). Therefore, the level of basal autophagy is comparable between the three cell lines, and does not show correlation with verteporfin sensitivity.
Verteporfin inhibits gemcitabine-induced autophagy in vitro
MCF-7 cells stably expressing EGFP-LC3 (26,27) have been used extensively to study autophagy by monitoring both EGFP-LC3 puncta and degradation. They showed a ~2.5-fold increase in punctate EGFP-LC3, representing autophagosomes, when treated with 500nM gemcitabine (Fig. 2A-B). Co-treatment with 10µM verteporfin considerably reduced gemcitabine-induced punctate EGFP-LC3 and increased diffuse cytoplasmic fluorescence (Fig. 2A-B). EGFP-LC3 is degraded upon autophagosome-lysosome fusion, but the EGFP moiety is more resistant to lysosomal degradation and transiently accumulates as a marker of increased autophagic flux (26). MCF-7 EGFP-LC3 cells exposed to gemcitabine for 24h showed a decrease in EGFP-LC3 levels and an increase in free EGFP, indicative of autophagy stimulation (Fig. 2C). Co-treatment with verteporfin prevented gemcitabine-induced EGFP accumulation, confirming that verteporfin inhibits gemcitabine-induced autophagy (Fig. 2C).
Inhibition of gemcitabine-induced autophagy by verteporfin. (A-C) MCF-7 EGFP-LC3 cells were exposed to 500nM gemcitabine (Gem) for 24h in the presence or absence of 10µM verteporfin, or to 0.1% DMSO vehicle control. Cells were fixed and stained with Hoechst 33342 and punctate EGFP-LC3 fluorescence was (A) visualized and (B) quantified using a Cellomics ArrayScan VTI automated fluorescence microscope. (* p < 0.01, Student's t-test)(mean ± S.D (error bars), n=3). (C) MCF-7 EGFP-LC3 cells were exposed to 0.1% DMSO, 500nM gemcitabine, or 30nM rapamycin, a known stimulator of autophagy, for 24h in the absence or presence of 10µM verteporfin. Lysates were collected and immunoblotted for EGFP. (D) BxPC-3 cells were exposed to 500nM gemcitabine for 24h in the presence or absence of 10µM verteporfin, 100nM bafilomycin A1, or 0.1% DMSO vehicle control. Lysates were collected and immunoblotted for LC3 and p62. β-tubulin was monitored as a loading control.
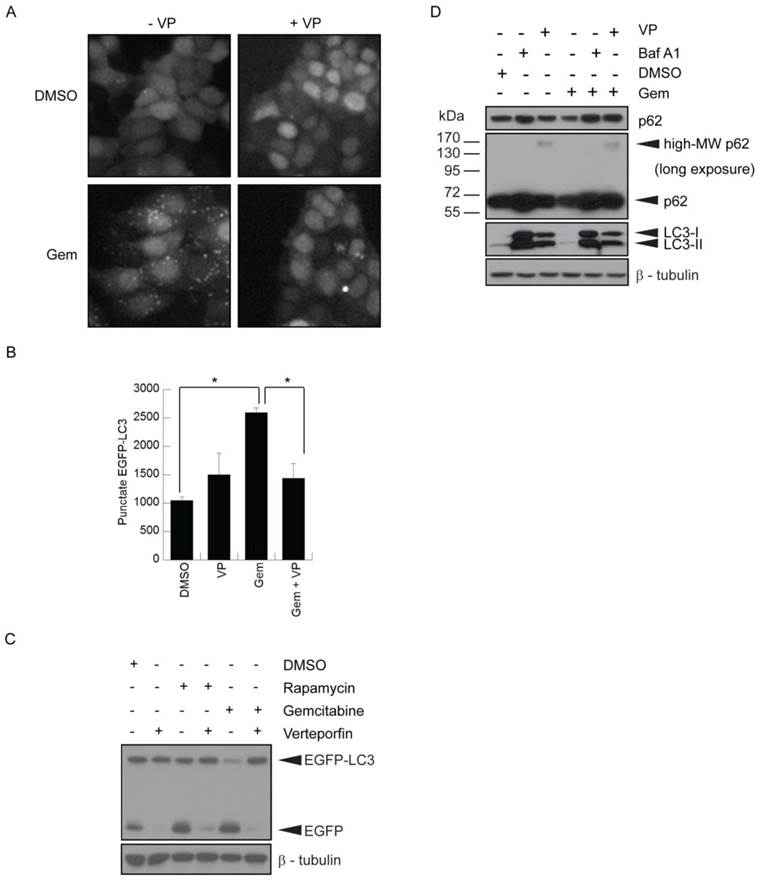
The effect of verteporfin on gemcitabine-induced autophagy was also demonstrated in BxPC-3 pancreatic cancer cells. Cells exposed to 500nM gemcitabine showed a decrease in p62 levels compared to the vehicle control (Fig. 2D). Co-treatment with 100nM bafilomycin A1 or 10µM verteporfin prevented gemcitabine-induced p62 degradation (Fig. 2D), confirming that verteporfin inhibits gemcitabine-induced autophagy in BxPC-3 cells. Additionally, we have observed that verteporfin induces the appearance of high-MW forms of p62 that are SDS-stable and resistant to denaturing conditions (Fig. 2D), and only does so at concentrations that inhibit autophagy, making it a useful marker of verteporfin activity (Fig. 2D and unpublished). Whether the high-MW p62 represents covalent adducts or other modifications is under investigation. Immunoblotting for LC3 in the same experiment showed that LC3-I and LC3-II accumulated in the presence of bafilomycin A1 and verteporfin, further illustrating that autophagy was inhibited in the presence of these drugs.
Effect of gemcitabine on BxPC-3 and SU86.86 proliferation
Exposure to 50-1000nM gemcitabine for 7 days significantly inhibited BxPC-3 and SU86.86 cell proliferation at doses as low as nM (Fig. 3A). Images taken throughout the assay showed few intact cells, evidence of gemcitabine cytotoxicity. Similar assays were carried out in the presence of verteporfin combined with gemcitabine, and no obvious enhancement of killing was observed (data not shown), due in part to the high potency of gemcitabine in vitro. However, many studies have shown large discrepancies between in vitro and in vivo effects of gemcitabine (16,38,39). Therefore we decided to proceed with a preliminary set of in vivo experiments using BxPC-3 and SU86.86 cells as subcutaneous xenografts in mice to examine gemcitabine sensitivity in vivo.
Effect of gemcitabine treatment on selected PDAC cell lines and tumour xenografts. (A) BxPC-3 and SU86.86 cells were treated with 50-1000nM gemcitabine (Gem) in vitro for up to 7 days. Media and drugs were replenished every third day. Cell viability and proliferation was measured as in Fig. 1A (mean ± S.D (error bars), n=4). (B) BxPC-3 and SU86.86 tumor-bearing mice were treated with saline as a control or with gemcitabine at 120mg/kg or 240mg/kg i.p. once per week over a period of four weeks. Treatment was initiated when tumors reached 100-150 mm3 (day 24 for BxPC-3 and day 25 for SU86.86). Tumor growth was monitored using digital callipers. Tumor growth is presented as relative tumor size, where tumor volumes were normalized with respect to the tumor volume of each mouse on the initial day of treatment and the average of each group was plotted. Arrows indicate the last day of treatment for each tumor model. (mean ± S.E.M (error bars), n=6 for all groups at the start of the study).
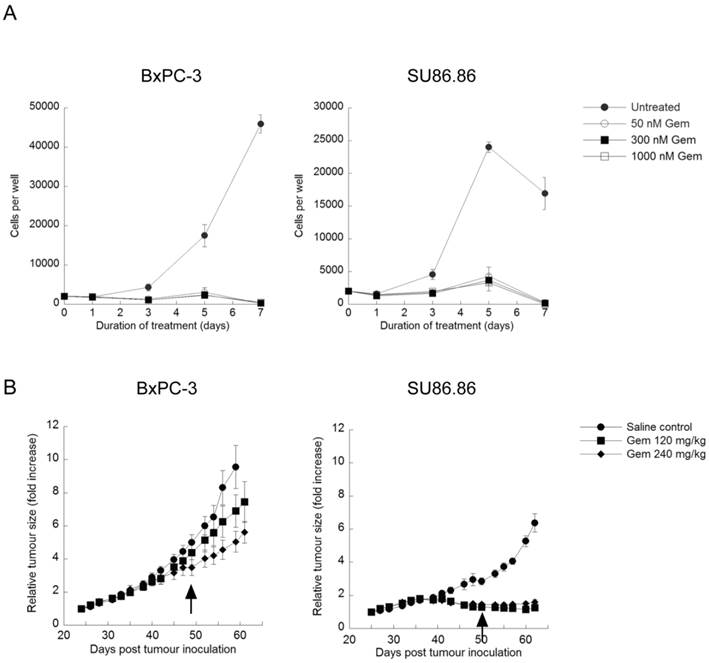
Rag2M mice bearing BxPC-3 or SU86.86 tumors were treated i.p. with gemcitabine at 120mg/kg or 240mg/kg once a week for 4 weeks (Q7Dx4) and tumor size was monitored. Animals with SU86.86 tumors were effectively treated with gemcitabine at the 120mg/kg dose; the tumor size in these mice remained unchanged over the course of these studies, 61 days post tumor inoculation, whereas tumors in the saline-treated control group exhibited a 6-fold increase in tumor volume (Fig. 3B). BxPC-3 tumors were much less sensitive to gemcitabine in vivo even though they appeared as sensitive to the drug in vitro. At the 120mg/kg gemcitabine dose there was a 7-fold increase in tumor volume compared with a 9-fold increase observed in saline control treated animals at the end of the study (Fig. 3B). When animals bearing established BxPC-3 tumors were treated with 240mg/kg gemcitabine there was a 5-fold increase in tumor volume (Fig. 3B). The more modest dose-dependent antitumor effects observed in BxPC-3 xenografts in response to gemcitabine treatment established it as a model suitable for investigating whether verteporfin can increase gemcitabine activity.
Verteporfin formulation and pharmacokinetics following a single administration
The in vitro studies suggest that inhibition of autophagy in a tumor will require µM levels of verteporfin over several days. Visudyne®, the commercial liposomal formulation of verteporfin developed for photodynamic therapy, is formulated at a very low verteporfin-to-lipid ratio (0.028mg/mg), limiting the highest dose that can be administered i.p. to about 20mg/kg. At this dose verteporfin tumor accumulation following i.v. administration was very poor, with tumor concentrations rapidly decreasing to 0.28µg/g (0.39µM) over 24h (unpublished). To increase verteporfin tumor accumulation we developed an alternate micellar formulation with DSPE-mPEG2000 as described in Methods and Materials.
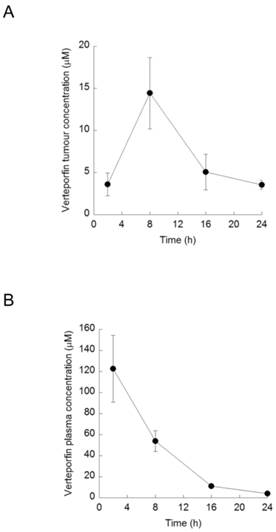
Rag2M mice bearing BxPC-3 tumors were treated with a single i.p. dose of verteporfin at 45mg/kg and tumors were harvested 2-24h later for UPLC-MS/MS analysis. Verteporfin tumor concentration peaked 8h post administration at 10.4µg/g (14.5µM, assuming 1g tumor tissue = 1ml), and remained > 2.6µg/g (3.5µM) for 24h (Fig. 4A). Plasma verteporfin concentration 2h post i.p. was 88.1µg/ml (122µM) and remained >2.8µg/ml (3.9µM) for 24h (Fig. 4B). Thus 45mg/kg verteporfin in the DSPEmPEG2000 formulation achieved tumor and plasma drug levels within the range that elicited anti-proliferative and autophagy-inhibiting effects in vitro.
Verteporfin accumulation in vivo. BxPC-3 tumor-bearing Rag2M mice were treated with verteporfin at 45mg/kg i.p. (A) Tumors and (B) blood samples were harvested 2, 8, 16, and 24h after administration for pharmacokinetic analysis. (mean ± S.D. (error bars), n=3).
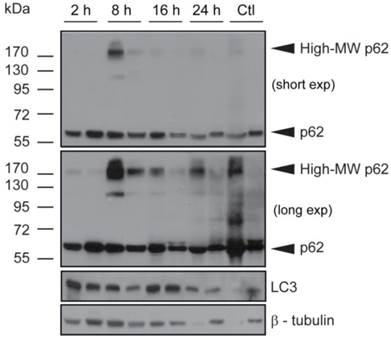
Verteporfin treatment induces high-MW p62 forms and LC3 accumulation in BxPC-3 tumor tissue in vivo
Tumors obtained from mice used for verteporfin pharmacokinetic analysis were also subjected to western blot analysis of p62 and LC3. The western blot from one control tumor (Fig. 5; Ctl left), showed an intense p62 smear and no LC3 or tubulin, suggesting its protein content was largely degraded during preparation; therefore, only the second control (Fig. 5; Ctl right) was considered for analysis. Additionally, while the control tumor showed only 60-kDa p62, small amounts of high-MW p62 were detected along with 60-kDa p62 2h after verteporfin administration (Fig.5), and increased dramatically 8h post administration, which correlated with the rise in verteporfin concentration in tumors (Fig. 4). Interestingly, the amount of high-MW p62 remained high until 24 h (Fig. 5) although the amount of verteporfin detected in tumors at 24h post administration had fallen to that observed at 2h (Fig. 4), implying that high-MW p62 remains in the tumor for some time after drug clearance (Fig. 5). Immunoblotting for LC3 showed that LC3 accumulated 2-16h post verteporfin administration (Fig. 5), similar to what was observed in cell culture, and providing evidence that autophagy was inhibited by verteporfin in vivo.
Effect of verteporfin on BxPC-3 tumour xenograft tissue. BxPC-3 tumors harvested 2, 8, 16, and 24h after a single verteporfin administration at 45mg/kg were also subjected to western blot analysis. Tumor sections were homogenized and immunoblotted for p62. In a separate experiment, the same lysates were probed for LC3. β-tubulin was monitored as a loading control for both experiments (n=2 per group).
Verteporfin moderately enhances the anti-tumor activity of gemcitabine in BxPC-3 xenografts
Rag2M mice bearing subcutaneous BxPC-3 tumors were dosed i.p. 3 times per week for 4 weeks with 45mg/kg verteporfin alone, or with gemcitabine i.p. once weekly for 4 weeks at 120mg/kg or 240mg/kg alone, or with both. Animals in the control group were treated with the delivery vehicle DSPE-mPEG2000 micelles at the same concentration and schedule as the verteporfin-loaded DSPE-mPEG2000. No toxicity was observed in any of the treatment groups and no data was excluded for analysis.
Treatment with verteporfin alone had minimal effect on tumor volume (Fig. 6, upper panels). For example, at day 57, one week after conclusion of treatment, tumors had increased 7.1-fold since the first day of treatment, compared to 8.4-fold in control mice. Treatment with 120mg/kg or 240mg/kg gemcitabine alone had a significant effect on tumor volume (Fig. 6, upper panels); at day 57, relative tumor volume increases were 5.5-fold and 6-fold, respectively (p < 0.05, days 53 and 55).
Anticancer efficacy of verteporfin, gemcitabine and the combination in in a BxPC-3 tumor model. BxPC-3 tumor-bearing Rag2M mice were given i.p. administrations of (A) a combination of 45mg/kg verteporfin and 120mg/kg gemcitabine or each drug on its own or (B) a combination of 45mg/kg verteporfin and 240mg/kg gemcitabine or each drug on its own for a period of four weeks. Treatment was initiated when tumors reached 100-150mm3, day 25. DSPE-PEG micelles without drug were delivered as the control. Tumor growth is presented as both average tumor volume (top) and relative tumor size (middle) as in Fig. 3B. Kaplan-Meier survival curves (bottom) illustrate the number of surviving mice in treatment group post drug administration. Arrows indicate the last day of treatment. (** p < 0.01)(mean ± S.E.M (error bars), n=8 for all groups at the start of the study).
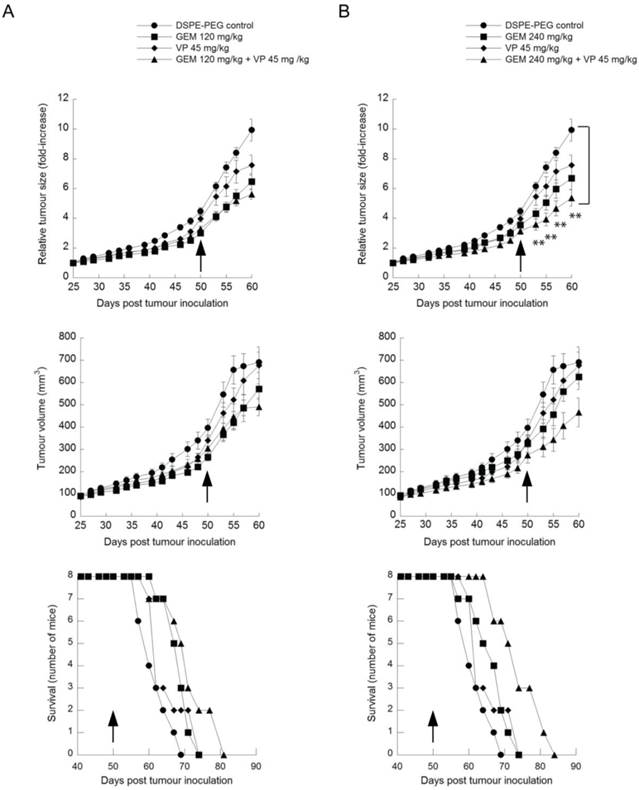
Kaplan-Meier survival curves were generated by monitoring the number of surviving mice after treatment using 800mm3 as the defined humane endpoint (Fig. 6, lower panels). None of the groups treated with a single agent showed a significant survival advantage over controls.
Mice treated with both verteporfin and 120mg/kg gemcitabine showed a relative tumor volume increase of 5.2-fold on day 57 (Fig. 6, upper left panel) that was not significantly different from gemcitabine alone. Mice treated with verteporfin and 240mg/kg gemcitabine showed a 4.7-fold increase, almost 50% less than that observed in the control group (Fig. 6, upper right panel); their tumors grew noticeably slower throughout the entire study, producing a highly significant effect on relative tumor growth compared to the control (p < 0.01, days 53 to 60). Overall the effect of combining verteporfin and 240mg/kg gemcitabine on relative tumor growth was additive compared to either treatment alone.
Comparing Kaplan-Meier survival curves, the arm representing the combined treatment regimen of verteporfin and 240mg/kg gemcitabine shifted to the right and did not overlap with any other treatment arm, depicting a convincing survival advantage over the control and all other treatments tested: a survival advantage of 14 days over vehicle control and 10 days over 240mg/kg gemcitabine alone (Fig. 6, lower right panel). In summary, combining verteporfin with 240mg/kg gemcitabine provided antitumor effects that were significantly better than what could be achieved with the drugs used as single agents.
Discussion
PDAC is a particularly aggressive cancer lacking effective therapeutic options (40). A recent publication describing PDAC as autophagy-dependent showed in vitro and in vivo PDAC growth was inhibited using the late-stage autophagy inhibitor CQ (13). This, in turn prompted initiation of clinical trials using HCQ. However, a critical review of preclinical studies investigating PDAC response to CQ reveals variable responses across cell lines and significant differences between in vitro and in vivo results (13,41). Clearly, the claim that PDAC is addicted to autophagy for proliferation appears to be more complex than first envisioned. Pharmacological autophagy inhibition using CQ and HCQ has been the subject of a number of combination therapy clinical trials with limited success (7,23). Issues that limit the therapeutic potential of HCQ clinically include the challenges of achieving autophagy-inhibiting drug levels in patients and the fact that prolonged accumulation of undigested autophagosomes can actually stimulate autophagy (23,24). In this study, we used verteporfin, an inhibitor of autophagosome formation, to explore autophagy inhibition as a strategy against PDAC when used alone and in combination with gemcitabine.
From a panel of 8 PDAC cell lines studied, only BxPC-3 and SU86.86 were highly sensitive to verteporfin treatment. These cells showed a clear dose response to the drug and 10µM verteporfin was sufficient to completely inhibit cell proliferation. The observation that a mechanistically distinct autophagy inhibitor produced a spectrum of autophagy-dependence responses warranted further study; however, it is recognized that the effects, like HCQ and CQ, are cell-line specific. Verteporfin was shown to inhibit gemcitabine-induced autophagy in both reporter MCF-7 EGFP-LC3 cells and BxPC-3 cells, providing rationale for us to explore the in vivo effects of verteporfin both on its own and in combination with gemcitabine.
Characterization of BxPC-3 and SU86.86 cell lines revealed both cell lines as extremely sensitive to gemcitabine in vitro, but that response was recapitulated in vivo only in the SU86.86 tumor model. BxPC-3 xenografts showed a moderate dose-dependent delay in tumor growth, but failed to cause tumor regression even at the highest dose (240mg/kg). This was in contrast to the in vitro data and illustrates one of the hurdles when trying to define effective new agents for use in treating pancreatic cancer (38,41,42). Regardless, the less responsive BxPC-3 xenograft model was better-suited for our in vivo study since it provided a therapeutic window for evaluating whether autophagy inhibition by verteporfin could enhance gemcitabine efficacy.
Verteporfin failed to show antitumor activity on mice bearing established BxPC-3 xenografts, despite evidence the drug accumulated at the tumor site for 18-20h after a single injection, achieving concentrations sufficient to affect cell proliferation in vitro. The generation of high-MW p62 and accumulation of LC3 in BxPC-3 tumor tissue by verteporfin confirmed in vivo autophagy inhibition within 24h of drug administration, but this was not enough to significantly inhibit tumor growth. In addition to our study, Mirzoeva et al recently reported negative antitumor responses to CQ using a BxPC-3 xenograft model despite encouraging in vitro results (41). Two clear issues have arisen with this therapeutic strategy: 1) it is apparent that PDAC dependence on autophagy is highly variable among cell lines; and 2) even extremely encouraging in vitro data are not necessarily indicative of in vivo success. Whether the latter is due to issues with drug delivery in vivo may be overcome by optimizing drug delivery and dosage; however, due to the large number of variables involved, targeting autophagy as a single-strategy modality seems an unlikely prospect for combating pancreatic cancer.
A more encouraging in vivo anticancer response to verteporfin was observed in combination with gemcitabine, which modestly inhibited tumor growth and conferred a survival advantage of ~10 days. These results support a cytoprotective role for gemcitabine-induced autophagy in contrast with other reports claiming autophagy contributes to tumoral cell death (17,18). This discrepancy is likely due to the fact that the aforementioned studies characterized the effects of inhibiting gemcitabine-induced autophagy only in cell culture and using 3-MA to inhibit autophagy, which has a number of other targets, and is unsuitable for in vivo use (43).
Studies showing dramatic tumor regression via autophagy suppression have largely been carried out using genetic inhibition (44,45), perhaps another reason for the spectrum of in vivo preclinical results published and the relatively minimal effects reported from clinical trials using CQ (23,46). The efficacy of autophagy inhibition in terms of increasing the potency of autophagy-inducing therapies is likely dependent on variables including genetic background, tumor microenvironment, and the therapeutic agent(s) used. It is therefore imperative to design rigorous in vivo studies and to report the results transparently and without bias (47) in an effort to successfully apply autophagy inhibition to the clinic.
The observed enhanced efficacy and survival advantage using verteporfin in the presence of gemcitabine reinforces the notion that gemcitabine-induced autophagy is a cytoprotective response and that verteporfin can sensitize PDAC to gemcitabine. It should be noted that none of the treatments showed tumor regression; however, in the context of pancreatic cancer, which has a 5-year survival rate of only 6% (10,48), any advancement in therapeutic efficacy should be considered valuable and worthy of further exploration.
Acknowledgements
The authors acknowledge the preclinical work aided by the Investigational Drug Program's Animal Care Team of Dana Masin, Dita Strutt, Maryam Osooly, Maria Rizza, and Hong Yan.
Grant Support: This work was funded by the Canadian Cancer Society and the Canadian Institutes of Health Research proof of principle grant 102196.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Mizushima N. Physiological functions of autophagy. Curr Top Microbiol Immunol. 2009;335:71-84
2. Weidberg H, Shvets E, Elazar Z. Biogenesis and cargo selectivity of autophagosomes. Ann Rev Biochem. 2011;80:125-56
3. Degenhardt K, Mathew R, Beaudoin B, Bray K, Anderson D, Chen G. et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell. 2006;10:51-64
4. Amaravadi RK, Lippincott-Schwartz J, Yin X-M, Weiss WA, Takebe N, Timmer W. et al. Principles and current strategies for targeting autophagy for cancer treatment. Clin Canc Res. 2011;17:654-66
5. Apel A, Herr I, Schwarz H, Rodemann HP, Mayer A. Blocked autophagy sensitizes resistant carcinoma cells to radiation therapy. Cancer Res. 2008;68:1485-94
6. Ertmer A, Huber V, Gilch S, Yoshimori T, Erfle V, Duyster J. et al. The anticancer drug imatinib induces cellular autophagy. Leukemia. 2007;21:936-42
7. Amaravadi RK, Yu D, Lum JJ, Bui T, Christophorou MA, Evan GI. et al. Autophagy inhibition enhances therapy-induced apoptosis in a Myc-induced model of lymphoma. J Clin Invest. 2007;117:326-36
8. Qadir MA, Kwok B, Dragowska WH, To KH, Le D, Bally MB. et al. Macroautophagy inhibition sensitizes tamoxifen-resistant breast cancer cells and enhances mitochondrial depolarization. Breast Cancer Res Treat. 2008;112:389-403
9. Sasaki K, Tsuno NH, Sunami E, Kawai K, Hongo K, Hiyoshi M. et al. Resistance of colon cancer to 5-fluorouracil may be overcome by combination with chloroquine, an in vivo study. Anticancer Drugs. 2012;23:675-82
10. Care STE, Chappell H. Canadian Cancer Statistics 2011. In: (ed.) Evans J. Statistics. Canadian Cancer Society. 2011:135
11. Réz G, Tóth S, Pálfia Z. Cellular autophagic capacity is highly increased in azaserine-induced premalignant atypical acinar nodule cells. Carcinogenesis. 1999;20:1893-8
12. Fujii S, Mitsunaga S, Yamazaki M, Hasebe T, Ishii G, Kojima M. et al. Autophagy is activated in pancreatic cancer cells and correlates with poor patient outcome. Cancer Sci. 2008;99:1813-9
13. Yang S, Wang X, Contino G, Liesa M, Sahin E, Ying H. et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 2011;25:717-29
14. Burris H, Moore M, Andersen J, Green M, Rothenberg M, Modiano M. et al. Improvements in survival and clinical benefit with gemcitabine as first- line therapy for patients with advanced pancreas cancer: a randomized trial. J Clin Oncol. 1997;15:2403-13
15. Wang X-Y, Li H-J, Wen H, Yan D, Peng S-Y. Influence of the adjuvant therapy on the survival of patients with stage II pancreatic carcinoma. Front Med China. 2010;4:430-5
16. Hung SW, Mody HR, Govindarajan R. Overcoming nucleoside analog chemoresistance of pancreatic cancer: a therapeutic challenge. Cancer Lett. 2012;320:138-49
17. Mukubou H, Tsujimura T, Sasaki R, Ku Y. The role of autophagy in the treatment of pancreatic cancer with gemcitabine and ionizing radiation. Int J Oncol. 2010;37:821-8
18. Pardo R, Lo Ré A, Archange C, Ropolo A, Papademetrio DL, Gonzalez CD. et al. Gemcitabine induces the VMP1-mediated autophagy pathway to promote apoptotic death in human pancreatic cancer cells. Pancreatology. 2010;10:19-26
19. Rikiishi H. Possible Role of Autophagy in the Treatment of Pancreatic Cancer with Histone Deacetylase Inhibitors. Cancers. 2010;2:2026-43
20. Schneider P, Korolenko TA, Busch U. A review of drug-induced lysosomal disorders of the liver in man and laboratory animals. Microsc Res Tech. 1997;36:253-75
21. McAfee Q, Zhang Z, Samanta A, Levi SM, Ma X-H, Piao S. et al. Autophagy inhibitor Lys05 has single-agent antitumor activity and reproduces the phenotype of a genetic autophagy deficiency. Proc Natl Acad Sci USA. 2012;109:8253-8
22. Yang ZJ, Chee CE, Huang S, Sinicrope F a. The role of autophagy in cancer: therapeutic implications. Mol Cancer Ther. 2011;10:1533-41
23. Bristol ML, Emery SM, Maycotte P, Thorburn A, Chakradeo S, Gewirtz DA. Autophagy inhibition for chemosensitization and radiosensitization in cancer: do the preclinical data support this therapeutic strategy? J Pharmacol Exp Ther. 2013;344:544-52
24. Juhász G. Interpretation of bafilomycin, pH neutralizing or protease inhibitor treatments in autophagic flux experiments: novel considerations. Autophagy. 2012;8:1875-6
25. Kimura N, Kumamoto T, Kawamura Y, Himeno T, Nakamura K-I, Ueyama H. et al. Expression of autophagy-associated genes in skeletal muscle: an experimental model of chloroquine-induced myopathy. Pathobiology. 2007;74:169-76
26. Donohue E, Tovey A, Vogl AW, Arns S, Sternberg E, Young RN. et al. Inhibition of autophagosome formation by the benzoporphyrin derivative verteporfin. J Biol Chem. 2011;286:7290-300
27. Balgi AD, Fonseca BD, Donohue E, Tsang TCF, Lajoie P, Proud CG. et al. Screen for chemical modulators of autophagy reveals novel therapeutic inhibitors of mTORC1 signaling. PloS One. 2009;4:e7124
28. Curman D, Cinel B, Williams DE, Rundle N, Block WD, Goodarzi AA. et al. Inhibition of the G2 DNA damage checkpoint and of protein kinases Chk1 and Chk2 by the marine sponge alkaloid debromohymenialdisine. J Biol Chem. 2001;276:17914-9
29. Deer EL, González-Hernández J, Coursen JD, Shea JE, Ngatia J, Scaife CL. et al. Phenotype and genotype of pancreatic cancer cell lines. Pancreas. 2010;39:425-35
30. Ling J, Kang Y, Zhao R, Xia Q, Lee D-F, Chang Z. et al. KrasG12D-induced IKK2/β/NF-κB activation by IL-1α and p62 feedforward loops is required for development of pancreatic ductal adenocarcinoma. Cancer Cell. 2012;21:105-20
31. Lock R, Roy S, Kenific CM, Su JS, Salas E, Ronen SM. et al. Autophagy facilitates glycolysis during Ras-mediated oncogenic transformation. Mol Biol Cell. 2011;22:165-78
32. Guo JY, Chen H-Y, Mathew R, Fan J, Strohecker AM, Karsli-Uzunbas G. et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 2011;25:460-70
33. Berrozpe G, Schaeffer J, Peinado MA, Real FX, Perucho M. Comparative analysis of mutations in the p53 and K-ras genes in pancreatic cancer. Int J Cancer. 1994;58:185-91
34. Sun C, Yamato T, Furukawa T, Ohnishi Y, Kijima H, Horii A. Characterization of the mutations of the K-ras, p53, p16, and SMAD4 genes in 15 human pancreatic cancer cell lines. Oncol Rep. 2001;8:89-92
35. Loukopoulos P, Kanetaka K, Takamura M, Shibata T, Sakamoto M, Hirohashi S. Orthotopic transplantation models of pancreatic adenocarcinoma derived from cell lines and primary tumors and displaying varying metastatic activity. Pancreas. 2004;29:193-203
36. Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T. et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO. 2000;19:5720-8
37. Klionsky DDJ, Abdalla FFC, Abeliovich H, Abraham RT, Abraham RT, Acevedo-Arozena A. et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2012;8:445-544
38. Rathos MJ, Joshi K, Khanwalkar H, Manohar SM, Joshi KS. Molecular evidence for increased antitumor activity of gemcitabine in combination with a cyclin-dependent kinase inhibitor, P276-00 in pancreatic cancers. J Transl Med. 2012;10:161
39. Long J, Zhang Y, Yu X, Yang J, LeBrun DG, Chen C. et al. Overcoming drug resistance in pancreatic cancer. Exp Opin Ther Tar. 2011;15:817-28
40. Kang R, Tang D. Autophagy in pancreatic cancer pathogenesis and treatment. Am J Cancer Res. 2012;2:383-96
41. Mirzoeva OK, Hann B, Hom YK, Debnath J, Aftab D, Shokat K. et al. Autophagy suppression promotes apoptotic cell death in response to inhibition of the PI3K-mTOR pathway in pancreatic adenocarcinoma. J Mol Med (Berl). 2011;89:877-89
42. Sloat BR, Sandoval MA, Li D, Chung W-G, Lansakara-P DSP, Proteau PJ. et al. In vitro and in vivo anti-tumor activities of a gemcitabine derivative carried by nanoparticles. Int J Pharm. 2011;409:278-88
43. Rubinsztein DC, Gestwicki JE, Murphy LO, Klionsky DJ. Potential therapeutic applications of autophagy. Nat Rev Drug Discov. 2007;6:304-12
44. Cufí S, Vazquez-Martin A, Oliveras-Ferraros C, Corominas-Faja B, Urruticoechea A, Martin-Castillo B. et al. Autophagy-related gene 12 (ATG12) is a novel determinant of primary resistance to HER2-targeted therapies: utility of transcriptome analysis of the autophagy interactome to guide breast cancer treatment. Oncotarget. 2012;3:1600-14
45. Hu Y-L, DeLay M, Jahangiri A, Molinaro AM, Rose SD, Carbonell WS. et al. Hypoxia-induced autophagy promotes tumor cell survival and adaptation to antiangiogenic treatment in glioblastoma. Cancer Res. 2012;72:1773-83
46. Chen N, Karantza V. Autophagy as a therapeutic target in cancer. Cancer Biol Ther. 2011;11:157-68
47. Landis SC, Amara SG, Asadullah K, Austin CP, Blumenstein R, Bradley EW. et al. A call for transparent reporting to optimize the predictive value of preclinical research. Nature. 2012;490:187-91
48. Hezel AF, Kimmelman AC, Stanger BZ, Bardeesy N, Depinho RA. Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev. 2006;20:1218-49
Author contact
![]() Corresponding author: Michel Roberge, Department of Biochemistry and Molecular Biology, University of British Columbia, 2350 Health Sciences Mall, Vancouver, B.C., CANADA, V6T 1Z3. Ph: 604.822.2304; Fax: 604.822.5227; Email michelrubc.ca
Corresponding author: Michel Roberge, Department of Biochemistry and Molecular Biology, University of British Columbia, 2350 Health Sciences Mall, Vancouver, B.C., CANADA, V6T 1Z3. Ph: 604.822.2304; Fax: 604.822.5227; Email michelrubc.ca