Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2013; 4(7):557-565. doi:10.7150/jca.6689 This issue Cite
Research Paper
Antitumor Activity of TAK-285, an Investigational, Non-Pgp Substrate HER2/EGFR Kinase Inhibitor, in Cultured Tumor Cells, Mouse and Rat Xenograft Tumors, and in an HER2-Positive Brain Metastasis Model
Akiko Nakayama, Shinji Takagi, Tadashi Yusa, Masahiro Yaguchi, Akira Hayashi, Toshiya Tamura, Youichi Kawakita, Tomoyasu Ishikawa, Yoshikazu Ohta ![]()
Takeda Pharmaceutical Company Ltd, Fujisawa, Kanagawa 251-8555 Japan.
Received 2013-5-14; Accepted 2013-7-19; Published 2013-8-16
Abstract
Breast cancer therapy has improved following the development of drugs with specific molecular targets, exemplified by inhibitors of human epidermal growth factor receptor-2 (HER2) or epidermal growth factor receptor (EGFR) such as trastuzumab and lapatinib. However, these drugs have little effect on brain metastasis due to the combined effects of poor penetration of the blood-brain barrier and their removal from the central nervous system (CNS) by the p-glycoprotein (Pgp) drug efflux pump. We investigated the effects of TAK-285, a novel, investigational, dual EGFR/HER2 inhibitor that has been shown to penetrate the CNS and has comparable inhibitory efficacy to lapatinib which is a known Pgp substrate. Tested against a panel of 96 kinases, TAK-285 showed specificity for inhibition of HER family kinases. Unlike lapatinib, TAK-285 is not a substrate for Pgp efflux. In mouse and rat xenograft tumor models, TAK-285 showed antitumor activity against cancers that expressed HER2 or EGFR. TAK-285 was as effective as lapatinib in antitumor activity in a mouse subcutaneous BT-474 breast cancer xenograft model. TAK-285 was examined in a model of breast cancer brain metastasis using direct intracranial injection of BT-474-derived luciferase-expressing cells and showed greater inhibition of brain tumor growth compared to animals treated with lapatinib. Our studies suggest that investigational drugs such as TAK-285 that have strong antitumor activity and are not Pgp substrates may be useful in the development of agents with the potential to treat brain metastases.
Keywords: TAK-285, brain metastases, dual HER2/EGFR kinase inhibitor, lapatinib, murine xenografts.
Introduction
The development of distant metastases negatively impacts quality of life and survival, and at least 10% of all cancer patients will develop brain metastasis (1). Breast cancer is one of the most common primary tumors to develop associated brain metastases (1, 2), and while recent progress in cancer therapy has improved control of primary breast tumors, methods to effectively treat brain metastases from breast cancer remain inadequate. Consequently, there remains an urgent need to develop new effective agents for these patients (2, 3).
Brain tumors, whether primary or metastatic, are difficult to treat; curative surgery and resection are often not viable options. Most antineoplastic agents have limited access to the central nervous system (CNS) following systemic administration. One of the primary reasons for restricted drug access to the CNS is the blood-brain barrier (BBB), which limits the exposure to the CNS and, therefore limits the effectiveness of pharmacological agents in neuronal tissue. The exact biological mechanism(s) by which the BBB limits drug exposure is not clear, but in addition to epithelial tight junctions, enhanced efflux of chemotherapeutic agents out of the CNS space by endothelial cell membrane transporters in brain blood vessels plays a primary role (2). Key drug efflux transporters operational in preserving the BBB are the P-glycoprotein (Pgp) transporter, which belongs to the ATP-binding cassette (ABC) transporter family, and the breast cancer resistance protein (BCRP) ABCG2 (4, 5). These transport systems are responsible for the rapid removal of therapeutic agents from the CNS following local or systemic administration and thus contribute to relatively ineffective concentrations in the brain that characterize many antineoplastic agents. Novel antineoplastic agents that are able to penetrate the CNS and that are not substrates for Pgp or BCRP efflux may be useful in the treatment of CNS tumors.
In breast cancer, overexpression of human epidermal growth factor receptor 2 (HER2), a cell membrane receptor tyrosine kinase, is often associated with advanced disease and poor prognosis. Also, 70-80% of metaplastic breast carcinomas overexpress the epidermal growth factor receptor (EGFR) (6). Preclinical and clinical evidence has shown that agents targeting HER2 activity, such as the HER2 inhibitor trastuzumab (Herceptin®), and the dual EGFR/HER2 inhibitor lapatinib (Tykerb®), can be effective breast cancer therapies (7); both of these agents are used clinically in the management of HER2-positive breast cancer. Trastuzumab, a monoclonal antibody against HER2, is a large molecule that, while effective for the treatment of systemic disease, does not cross the BBB and is considered an ineffective treatment for HER2-positive brain tumors (8). Similarly, small molecular weight lapatinib, a 4-anilinoquinazoline kinase inhibitor, poorly distributes to the brain, is a known substrate for efflux by Pgp, and has limited efficacy in the treatment of HER2-positive brain tumors (8‒10).
TAK-285 is a novel, investigational, small molecule pyrrolo[3,2-d]pyrimidine-derived EGFR inhibitor that also specifically targets HER2 (11, 12), like lapatinib. TAK-285 has undergone clinical evaluation in advanced cancers (13, 14). Preclinical studies recently suggested that TAK-285 is a poor substrate for Pgp and other transporters responsible for maintaining the BBB (15, 16). Furthermore, preclinical in vivo studies have revealed that TAK-285 is distributed more efficiently to brain tissues than other HER2-targeted agents (16). These observations suggest that HER2/EGFR inhibitors that are not Pgp substrates may be useful in the development of agents for the treatment of patients with HER2/EGFR-positive brain tumors.
In the present studies, we confirmed that TAK-285 is not a substrate for Pgp in a cell line engineered to overexpress this transporter, examined the target specificity of TAK-285 and its efficacy against lung and colon tumor cells in vitro and in in vivo xenografts, and assessed the efficacy of TAK-285 in a model of breast cancer brain metastasis using breast cancer cells constitutively expressing luciferase (BTLUC) that allowed in vivo monitoring of tumor growth.
MATERIALS AND METHODS
Reagents
TAK-285 and lapatinib were synthesized as previously described (12, 18, 19). The identity of the synthesized material was confirmed using 1H nuclear magnetic resonance spectrum data and X-ray powder diffraction patterns; purity (~99%) was determined by high-performance liquid chromatography analysis. Medium and liquid supplements for cell culture studies were purchased from GIBCO (Osaka, Japan). Rhodamine123 was purchased from Millipore (Billerica, MA), and luciferin was purchased from the WAKO Chemical Company (Osaka, Japan). All other chemicals were obtained from the WAKO Chemical Company. Primary antibodies used in Western blot analyses were anti-HER2 #2242, anti-phospho-HER2 #2244, anti-phospho-Akt #9271 and anti-phospho-MAPK #9101 (CellSignaling Technology, Boston, MA), anti-phospho-EGFR #05-483 (Upstate/MerckMillipore, Billerica, MA).
Cells and cell growth conditions
The BT-474 human breast cancer cell line was purchased from American Type Culture Collection (ATCC, Manassas, VA) and cultured in RPMI-1640 medium containing 10% fetal bovine serum (FBS). MES-SA human uterine sarcoma cells and multidrug-resistant MES-SA/DX-5 cells (20) were provided by Dr. Ron de Jong (Takeda San Diego, Inc., San Diego, CA) and cultured in McCoy's 5A medium containing 10% FBS. All cell cultures were maintained at 37ºC in a humidified incubator in an atmosphere of 5% CO2.
Some experiments utilized BT-474 cells transfected to stably express luciferase. To generate the luciferase expression vector, the luciferase/neomycin domain of the pGL4.17[luc2/Neo] Vector E6721 (Promega, Madison, WI) and cytomegalovirus (CMV) promoter domain of the pGL4.75[hRluc/CMV] Vector E6931 (Promega) were combined and this construct was transfected into the BT-474 cell line using Lipofectamine-Plus™ Transfection Reagent (Life technologies, Carlsbad, CA). Clones stably expressing luciferase were selected in neomycin and those possessing the highest luciferase expression (designated BTLUC) were expanded, cryopreserved, and used in the studies described below.
A-431, NCI-N87, SK-BR-3, Calu-3, MDA-MB-453, and MRC-5 cells were obtained from ATCC and maintained according to the supplier's specifications in the appropriate cell culture growth medium supplemented with 10% FBS.
Animals
Female BALB/c nu/nu mice and female F344/N athymic (rnu/rnu) rats were purchased from CLEA Japan (Tokyo, Japan). Animals were maintained under controlled conditions (23 ± 2ºC room temperature, 55 ± 10% humidity and 12 h/12 h dark/light) and had food and water available ad libitum. All experimental procedures were approved by the Institutional Animal Care and Use Committee at Takeda Pharmaceutical Company Ltd.
In vitro methods Measurement of kinase activity
HER2 and EGFR kinase assays were performed as previously described (11, 12). Inhibitory activity of TAK-285 was assessed against a panel of 96 kinases (KinaseProfiler, Millipore, MA).
Assessment of Pgp function
To determine if TAK-285 was a substrate for Pgp, MES-SA and MES-SA/Dx-5 cells were seeded at 106 cells/well in 6-well plates and incubated in growth media overnight. The next day, cells were washed and incubated in serum-free McCoy's 5A medium containing 1 μmol/L rhodamine123 and either TAK-285 or lapatinib (10 μmol/L) for 1.5 h. Cells were then washed with serum-free medium, incubated for an additional 1.5 h in serum-free medium, and detached by trypsinization. Cellular luminescence was measured by FACSCalibur™ Flow Cytometry and analyzed by CellQuest™ (Becton Dickinson, Franklin Lakes, NJ) and FLOWJO software (Tree Star, Inc., Ashland, OR) as previously described (11, 12, 15).
Western blot analysis
Cells were seeded at 1 x 105 cells/well in 48-well plates. After incubation, the culture medium was discarded, cells were harvested by trypsinization, and cellular proteins were extracted by dissolution in 100 μL of 2 x sample buffer (BioRad, Hercules, CA). Proteins were denatured by boiling for 5 min. Total cellular proteins contained in a 10 μL aliquot were resolved by sodium dodecyl sulfate (SDS) polyacrylamide gel electrophoresis and proteins of interest were identified by a conventional chemiluminescent immunoblotting techniques using a LAS-1000 luminescence detector (Fuji Film, Tokyo, Japan).
Cell growth assays
BT-474, A-431, NCI-N87, SK-BR-3, Calu-3, MDA-MB-453, and MRC-5 cells were seeded at 3‒30 x 103 cells/well in 48 or 96 -well plates and incubated in growth medium. After 24 h, TAK-285 in dimethylsulfoxide (DMSO) was added to the medium to achieve final concentrations of 0.00097-25 µmol/L. After 3-7 days, cells were harvested following trypsin digestion and counted using a CDA-500 cell counter (Sysmex, Kobe, Japan) or using Cell Counting Kit-8 (Dojindo Laboratories, Kumamoto, Japan) and compared with DMSO controls. IC50 values and 95% confidence intervals (CI) were determined by logistic regression analysis (SAS software, SAS Institute, Cary, NC) from cell survival curves.
Cell signal inhibition assays
For cell signal inhibition assays, BT-474 cells were seeded at 1 x 105 cells/well in 48-well plates and incubated in growth medium overnight. On the following day, TAK-285 was added to the medium to achieve final concentrations of 6.25, 12.5, 25, 50, 100 nmol/L. After incubation for 2 h, cells were dissolved in SDS sample buffer and assayed by Western blot for the presence of phosphorylated HER2, Akt and MAPK compared with DMSO controls. A-431 cells were prepared in a similar fashion and incubated with TAK-285 12.5, 25, 50, 100, 200 nmol/L plus EGF 100 ng/mL and assayed by Western blot for phosphorylated EGFR. IC50 values and 95% CI were determined by logistic regression analysis of Western blot results.
In vivo methods Antitumor activity in xenograft models
In vivo antitumor efficacy was examined in nude mice or rats implanted subcutaneously with BT-474 cells or 4-1ST tumors, both of which express elevated levels of HER2, or with A-431 cells, which overexpress EGFR. BT-474 cells were injected in 6-week-old female BALB/c athymic (nu+/nu+) mice at 107 cells/animal in a 200 µL mixture (1:1) of Hank's buffered saline solution (HBSS) and Matrigel (Becton, Dickinson and Company, Franklin Lakes, NJ). A-431 cells were injected in 10-week-old female F344/N athymic (rnu/rnu) rats at 2 x 106 cells/animal in a 200 µL mixture (1:1) of HBSS and Matrigel. Human gastric tumor, 4-1ST was obtained from the Central Institute for Experimental Animals (Kanagawa, Japan) and maintained as an xenograft in BALB/c nu/nu nude mice. Minced 8 mm3 pieces of the 4-1ST tumors were injected by needle trocar into 9-week-old female BALB/c athymic (nu+/nu+) mice or 6-week-old female F344/N athymic (rnu/rnu) rats. When tumors reached 200‒300 mm3 in size, TAK-285 was emulsified in 0.5% methyl cellulose and administered orally twice daily (BID) for 2 weeks. Control animals received 0.5% methyl cellulose following the same schedule. Tumor volume was assessed every 2‒4 days during the dosing period using the formula tumor volume (mm3) = 1/2 x (long axis) x (short axis)2 as previously described (12). On the final day, treated/control values (T/C [%]) were calculated and statistically analyzed using Williams' test.
Intracranial xenograft induction and treatment
BTLUC cells were cultured in RPMI-1640 containing 10% FBS, harvested in logarithmic growth, and suspended at a density of 12.5 x 107 cells/mL in HBSS at room temperature. For intracranial injection, mice were anesthetized, placed in a stereotaxic restraint SR-5R (Narishige Group, Tokyo, Japan), and a small surgical incision was made in the skin covering the skull 2 mm to the right of the bregma. Cell suspensions (4 μL) containing approximately 5 x 105 BTLUC cells at room temperature were slowly (~8 seconds) injected intracranially at 3.5 mm below the skull surface using a 29-gauge needle. Immediately thereafter the incision was closed with surgical adhesive and the animals were monitored until conscious and returned to their cages.
Intracranial xenograft growth was assessed twice weekly by measuring luciferase-dependent luminescence using the IVIS Spectrum system (Xenogen, Alameda, CA). To determine luminescence, mice were administered luciferin by intraperitoneal injection (15 mg/mL phosphate-buffered saline, 1 mL/kg) and after 10 min, luminescence in each animal was determined. When the average luminescence intensity in xenograft-bearing mice reached 5 x 108 - 5 x 109 photons/sec, the mice were grouped for equal-sized tumors and drug treatment was initiated.
To confirm the presence of intracranial xenografts histologically, mice were euthanized 45 days after tumor cell injection, and the whole brain was removed and fixed in neutral buffered 10% formaldehyde solution overnight. The next day, tissue was dehydrated in 70% ethanol. Thereafter, paraffin-embedded tissues were sectioned into 4 μm slices, transferred to glass slides, dried, and stained using hematoxylin and eosin.
To determine the effect of TAK-285 or lapatinib on intracranial xenograft growth, these agents were emulsified in 0.5% methyl cellulose and administered orally (10 mL/kg) at 200 mg/kg BID daily for 3 weeks (1 week = 1 cycle). Control animals received 0.5% methyl cellulose alone following the same schedule. Luciferase-dependent luminescence was determined every 5-7 days. On the final day, treated/control values (T/C [%] = [Luminescencetreated / Luminescencecontrol] x 100) were calculated and statistically analyzed using Dunnett's test (21).
RESULTS
Selectivity of TAK-285
TAK-285 demonstrated inhibitory activity against HER2 and EGFR kinases with IC50 values for HER2 and EGFR of 17 nmol/L (95% CI 12-24) and 23 nmol/L (95% CI 18-30), respectively. In a panel of human kinases, TAK-285 showed weak inhibition (0‒50%) against 88 of 96 kinases tested, moderate inhibition (50-80%) against EGFR (L858R) and alkaline phosphatase, and strong inhibition (80‒100%) against HER family kinases including EGFR(L861Q) as well as against CDK3/cyclin E and Flt3 (Figure 1).
Selectivity of TAK-285 (1 μM) kinase inhibitory activity.

Pgp substrate activity
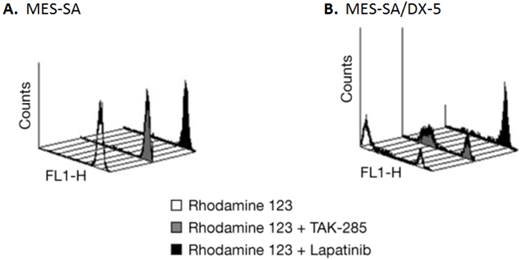
Lapatinib is a Pgp substrate and thus would not be a candidate for the treatment of brain tumors (22). To confirm whether TAK-285 is a substrate for the Pgp drug efflux system, the effect of TAK-285 and, for comparison, lapatinib, on rhodamine123 uptake and efflux was assessed in MES-SA human uterine sarcoma cells and a Pgp overexpressing sub-line, MES-SA/DX-5, by fluorescence-activated cell sorting analysis. As shown in Figure 2, parental MES-SA cells efficiently accumulated rhodamine123, demonstrated by the bright single peak (Figure 2A), while MES-SA/DX-5 cells showed two peaks of cellular luminescence (Figure 2B) reflecting rhodamine efflux mediated by Pgp. In MES-SA/DX-5 cells, incubation with TAK-285 resulted in little change in rhodamine-associated cellular luminescence while exposure to lapatinib appeared to inhibit rhodamine efflux. We obtained similar results using an alternative commercially available assay (15) that directly measured the ATPase activity of Pgp under cell-free conditions following TAK-285 or lapatinib exposure (data not shown). Lapatinib is a known substrate of the Pgp transport system (22) and our experiments confirm this and suggest that TAK-285 is not a Pgp substrate.
Pgp efflux assessment in MES-SA and MES-SA/DX-5 cells by rhodamine 123 uptake. A: rhodamine 123 uptake, but no efflux, in MES-SA cells. B: rhodamine 123 uptake and efflux in MES-SA/DX-5 cells that overexpress Pgp; effects of incubation with TAK-285 or lapatinib.
Cell growth inhibition assays
Sensitivities of various cell lines to growth inhibition by TAK-285 are shown in Table 1. In general, cells with higher expression of HER2 were more sensitive to TAK-285, but A-431 cells, which overexpress EGFR, were also sensitive to TAK-285. The IC50 for BT-474 cells was 0.017 µmol/L compared with 1.1 and 20 µmol/L in A-431 and MRC-5 cells which do not overexpress HER2. In other experiments, TAK-285 inhibited HER2 phosphorylation in BT-474 cells with IC50 values of 0.0093 µmol/L (95% CI 0.0065, 0.012). TAK-285 also inhibited Akt and MAPK phosphorylation in a dose-dependent manner, with IC50 values of 0.015 µmol/L (95% CI: 0.011, 0.018) and <0.0063 µmol/L, respectively. In A-431 cells, the IC50 value for TAK-285 inhibition of EGFR phosphorylation was 0.053 µmol/L (95% CI 0.040, 0.069).
Growth inhibitory effect of TAK-285 in cell lines.
| Cell lines | Growth inhibition (n=3) IC50, µmol/L (95% CI) | |
|---|---|---|
| HER2 overexpression | ||
| BT-474 | 0.017 | (0.012, 0.024) |
| NCI-N87 | 0.077 | (0.072, 0.083) |
| SK-BR-3 | 0.11 | (0.096, 0.13) |
| Calu-3 | 0.47 | (0.42, 0.53) |
| MDA-MB-453 | 0.91 | (0.76, 1.1) |
| EGFR overexpression | ||
| A-431 | 1.1 | (0.74, 1.5) |
| HER2 and EGFR normal expression | ||
| MRC-5 | 20.0 | (18, 21) |
TAK-285 and lapatinib in vivo activity
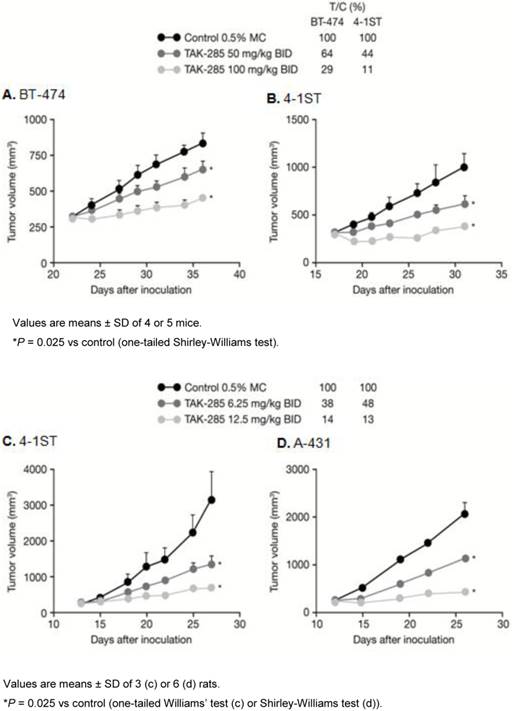
TAK-285 in vivo antitumor activity in murine xenograft models is shown in Figure 3. TAK-285 (100 mg/kg BID) inhibited BT-474 breast tumors and 4-1ST gastric tumor growth with T/C values of 29% and 11%, respectively, compared to controls (Figure 3A, B). Similarly, in rat xenografts, TAK-285 inhibited growth of tumors that overexpressed HER2 (4-1ST) or EGFR (A-431) with T/C values of 14% and 13%, respectively, at a dose of 12.5 mg/kg BID (Figure 3C, D). In other experiments (not shown), the pharmacokinetic profile for TAK-285 showed much greater drug exposure in rats compared with mice, thus requiring lower doses in the rat xenograft model.
Antitumor activity of TAK-285. A, B: in mouse and C, D: rat xenograft models.
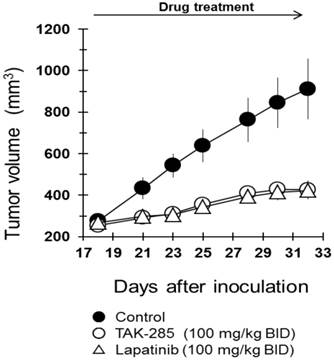
TAK-285 has been shown to effectively inhibit proliferation and viability of BT-474 cells with an IC50 of 17 nmol/L (Table 1), and under these in vitro conditions, possessed comparable cytotoxicity to lapatinib (IC50 ~10 nmol/L) (25). Consequently, studies were initiated to assess the in vivo activity of TAK-285 and lapatinib in mice bearing subcutaneous xenografts of BT474 human breast cancer cells that over-express HER2. In these studies, TAK-285 effectively inhibited subcutaneous xenograft growth when administered at a dose of 100 mg/kg BID, and after 2 weeks of treatment, the T/C ratio was 27% (Figure 4) and lapatinib at the same dose showed similar efficacy in this subcutaneous human tumor xenograft model system. Both treatment regimens were well tolerated, and there was no treatment-related mortality.
Antitumor efficacy of TAK-285 and lapatinib in murine subcutaneous xenografts. Inoculation of tumor on day 0 and drug treatment began on day 18.
Brain metastasis model
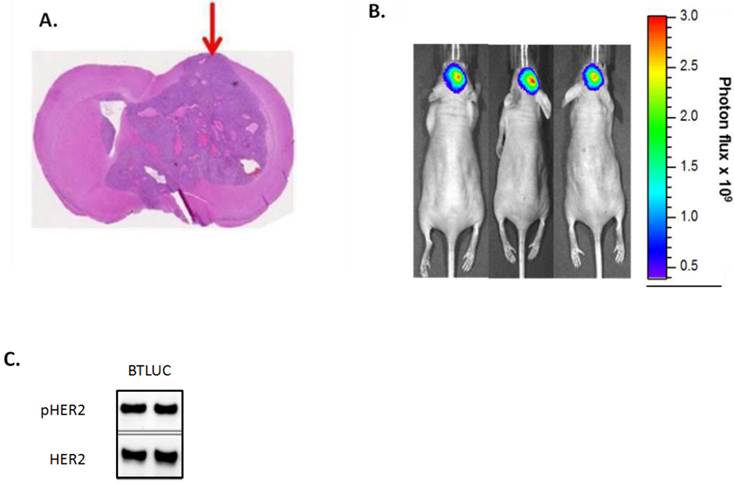
The tumor growth inhibitory effects of TAK-285 and lapatinib were tested in a murine intracranial injection model of brain metastases using BT-474-derived cerebral xenografts that resulted in highly reproducible tumor growth in a defined intracranial region (Figure 5A). For these experiments, we developed a line of luciferase-detectable BT-474 cells (BTLUC) that allowed non-invasive monitoring of intracranial xenograft growth (Figure 5B). BTLUC cells possessed highly activated HER2 with phosphorylation comparable to parental BT-474 cells (Figure 5C) and sensitivity to TAK-285 with IC50 value of 90 nmol/L (95% CI: 76-107nmol/L).
Intracranially injected BTLUC as a brain metastasis model and efficacy of TAK-285. A: representative nude mouse brain cross section showing brain tumor after 45 days. Arrow indicates injection site. B: in vivo fluorescence imaging of BTLUC brain tumors in nude mouse xenograft model 41 days after intracranial injection. Color intensity indicates photon flux measurements. C: HER2 activity in BTLUC cells assessed by Western blot.
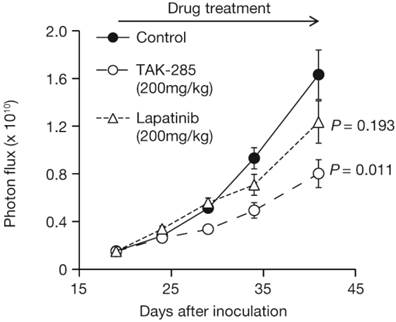
TAK-285 inhibited intracranial xenograft growth with T/C values of 44% on day 41 (Figure 6, P = 0.011 vs control). The T/C value for lapatinib on day 41 was 73%, was not significantly different from control-treated animals (P = 0.193) and was less effective than TAK-285.
Growth inhibition of intracranially-injected BTLUC brain metastasis xenografts by TAK-285 and lapatinib at 200 mg/kg BID orally for 3 weeks. P values are vs. control treatment.
DISCUSSION
The results of these studies demonstrate that TAK-285 showed high specificity for HER2 and EGFR kinase inhibition with IC50 values in low nanomolar concentrations. TAK-285 showed greater inhibition of cell growth in cell lines that overexpressed HER2, and also inhibited growth in cell lines that overexpressed EGFR. In mouse and rat xenograft tumor models, TAK-285 demonstrated antitumor activity in tumors that overexpress HER2 (BT-474, 4-1ST) or EGFR (A-431).
Few chemotherapeutic or molecularly targeted antineoplastic agents have shown efficacy in brain tumors, primarily due to the poor distribution of these agents into the brain, where the BBB prevents access and/or the Pgp efflux removes them from the CNS space. TAK-285 is not a substrate for the ABC-transporter or BCRP, and has been shown to be efficiently distributed into the brain (15). These results support those of Wu et al. (16, 17) who employed the more conventional Caco cell model system and found that, in contrast to lapatinib, TAK-285 is not a substrate for conventional drug efflux processes (16) and is not a substrate for conventional transport systems in the BBB. In vivo studies in rats have shown greater brain penetration (brain AUC/plasma AUC) with TAK-285, compared with lapatinib, a HER2 inhibitor, which is a substrate for Pgp (16).
In preclinical models of HER2-positive brain tumors, intracranial tumor cell injection was found to be a reproducible method to seed tumor cells, and xenografts developed in 2-3 weeks. In this model, TAK-285 was more effective than lapatinib, presumably due to differences in the ability of the two drugs to access the CNS. This implied that the BBB in this model was at least partially intact, and that TAK-285 was not being rapidly removed from the CNS and was not a substrate for drug efflux processes. In line with these results, a recent phase 1 clinical study in cancer patients revealed that TAK-285 is distributed into the CNS space after systemic administration (14). Our studies may provide useful models for the evaluation of similar compounds that achieve higher CNS concentrations.
The precise molecular mechanism that results in the development of brain-trophic metastasis is not known (23, 24). Our preliminary gene expression analysis in cell lines obtained by repeated selection cycles of brain metastasis revealed several candidate genes (not shown) that have not been reported previously (23). Further studies on gene regulation may prove to be of importance in the search for clinical markers for prediction of brain metastasis.
In conclusion, TAK-285 showed strong and selective dual kinase inhibition of HER2 and EGFR. TAK-285 inhibited the growth of a variety of malignant cell types and demonstrated antitumor activity in mouse and rat xenograft models. TAK-285 is not a Pgp substrate, and agents like TAK-285 that are not Pgp substrates may be useful in the development of drugs that have antitumor activity in brain metastasis in addition to extra-cranial tumors. Our studies reported here also suggest that this BTLUC brain metastasis model may be useful for the preclinical evaluation of investigational agents such as TAK-285, or agents derived from TAK-285 (25), that may have access to the CNS.
Abbreviations
ABC: ATP-binding cassette; AUC: area under the curve: BBB: blood-brain barrier; BCRP: breast cancer resistance protein; BID: twice daily; BTLUC: BT-474-derived luciferase-expressing; CI: confidence interval; CMV: cytomegalovirus; CNS: central nervous system; DMSO: dimethylsulfoxide; EGFR: epidermal growth factor receptor; FBS: fetal bovine serum; HBSS: Hank's buffered saline solution; HER2: human epidermal growth factor receptor-2; Pgp: p-glycoprotein; SDS: sodium dodecyl sulfate; T/C: treated/control values.
Acknowledgements
We would like to thank Dr. Shuichi Furuya for advice and guidance with this research. The authors would also like to acknowledge Dr. Kouki Hikami for the construction of the luciferase expression vector and the writing assistance of Jim Darnowski of Millennium: the Takeda Oncology Company and Stephen Mosley of FireKite Ltd, in the development of this manuscript, which was funded by Millennium.
Conflict of Interest
All authors are employees of Takeda Pharmaceutical Company Ltd, and this work was wholly supported by Takeda Pharmaceutical Company Ltd, Fujisawa, Japan.
References
1. Santarelli JG, Sarkissian V, Hou LC. et al. Molecular events of brain metastasis. Neurosurg Focus. 2007;22(3):E1
2. Steeg PS, Camphausen KA, Smith QR. Brain metastases as preventive and therapeutic targets. Nat Rev Cancer. 2011;11(5):352-63
3. Stemmler HJ, Heinemann V. Central nervous system metastases in HER-2-overexpressing metastatic breast cancer: A treatment challenge. Oncologist. 2008;13(7):739-50
4. Sun H, Dai H, Shaik N. et al. Drug efflux transporters in the CNS. Adv Drug Deliv Rev. 2003;55(1):83-105
5. Lin JH. How significant is the role of P-glycoprotein in drug absorption and brain uptake? Drugs Today (Barc). 2004;40(1):5-22
6. Reis-Filho JS, Milanezi F, Carvalho S. et al. Metaplastic breast carcinomas exhibit EGFR, but not HER2, gene amplification and overexpression: immunohistochemical and chromogenic in situ hybridization analysis. Breast Cancer Res. 2005;7(6):R1028-35
7. Hynes NE, Lane HA. ERBB receptors and cancer: the complexity of targeted inhibitors. Nat Rev Cancer. 2005;5(5):341-54
8. Gori S, Rimondini S, De Angelis V. et al. Central nervous system metastases in HER-2 positive metastatic breast cancer patients treated with trastuzumab: incidence, survival, and risk factors. Oncologist. 2007;12(7):766-73
9. Polli JW, Humphreys JE, Harmon KA. et al. The role of efflux and uptake transporters in [N-{3-chloro-4-[(3-fluorobenzyl)oxy]phenyl}-6-[5-({[2-(methylsulfonyl)ethyl]amino}methyl)-2-furyl]-4-quinazolinamine (GW572016, lapatinib) disposition and drug interactions. Drug Metab Dispos. 2008;36(4):695-701
10. Polli JW, Olson KL, Chism JP. et al. An unexpected synergist role of P-glycoprotein and breast cancer resistance protein on the central nervous system penetration of the tyrosine kinase inhibitor lapatinib (N-{3-chloro-4-[(3-fluorobenzyl)oxy]phenyl}-6-[5-({[2-(methylsulfonyl)ethyl]amino}methyl)-2-furyl]-4-quinazolinamine; GW572016). Drug Metab Dispos. 2009;37(2):439-42
11. Ishikawa T, Seto M, Banno H. et al. Design and synthesis of novel human epidermal growth factor receptor 2 (HER2)/epidermal growth factor receptor (EGFR) dual inhibitors bearing a pyrrolo[3,2-d]pyrimidine scaffold. J Med Chem. 2011;54(23):8030-50
12. Aertgeerts K, Skene R, Yano J. et al. Structural analysis of the mechanism of inhibition and allosteric activation of the kinase domain of HER2 protein. J Biol Chem. 2011;286(21):18756-65
13. LoRusso P, Chiorean EG, Heath E. et al. Phase 1 Dose-Escalation Study of the Investigational HER2/EGFR Inhibitor TAK-285 in Patients with Advanced Cancer. Cancer Research. 2011;70:P3-14 -20
14. Chiorean EG, Sausville EA, Heath EI. et al. Phase I study of TAK-285, an investigational HER2/EGFR inhibitor, in patients (pts) with advanced cancer: Updated results and assessment of human CSF distribution. J Clin Oncol. 2011;29(Suppl 15):Abstract2538
15. Tamura T, Takagi S, Horikoshi K. et al. Combined antitumor efficacies of TAK-285, a novel ErbB1/ErbB2 dual kinase inhibitor, with other anticancer drugs. Eur J Cancer Suppl. 2008 6(12, Abstract 314): 100-1
16. Wu JT, Liao M, Gordon J. et al. TAK-285, a Novel HER2/EGFR Inhibitor, Penetrates the CNS in Rats with an Intact Blood Brain Barrier (BBB). Cancer Research. 2009;69(Suppl 3):Abstract5098
17. Erdo F, Gordon J, Wu JT, Sziráki I. Verification of brain penetration of the unbound fraction of a novel HER2/EGFR dual kinase inhibitor (TAK-285) by microdialysis in rats. Brain Res Bull. 2012;87(4-5):413-9
18. Mcclure MS, Osterhout MH, Roschangar F. et al. A novel process for the preparation of lapatinib and its pharmaceutically acceptable salts. PCT Int; WO 02/02552. 2002
19. Tsou HR, Overbeek-Klumpers EG, Hallett WA. et al. Optimization of 6,7-Disubstituted-4-(arylamino)quinoline-3-carbonitriles as Orally Active, Irreversible Inhibitors of Human Epidermal Growth Factor Receptor-2 Kinase Activity. J Med Chem. 2005;48(4):1107-31
20. Harker WG, MacKintosh FR, Sikic BI. Development and characterization of a human sarcoma cell line, MES-SA, sensitive to multiple drugs. Cancer Res. 1983;43(10):4943-50
21. Dunnett CW, Tamhane AC. Some new multiple-test procedures for dose finding. J Biopharm Stat. 1998;8(3):353-66
22. GlaxoSmithKline. Lapatinib (Tykerb®) [Package Insert]. Research Triangle Park, NC: GlaxoSmithKline. 2012
23. Palmieri D, Bronder JL, Herring JM. et al. Her-2 Overexpression increases the metastatic outgrowth of breast cancer cells in the brain. Cancer Res. 2007;67(9):4190-98
24. Bos PD, Zhang XH, Nadal C. et al. Genes that mediate breast cancer metastasis to the brain. Nature. 2009;459(7249):1005-09
25. Downey T. Analysis of a multifactor microarray study using Partek genomics solution. Methods Enzymol. 2006;411:256-70
Author contact
![]() Corresponding author: Yoshikazu Ohta, PhD. Address: Takeda Pharmaceutical Company Ltd, Fujisawa, Kanagawa 251-8555 Japan. Tel:+81-466-32-2621 Fax:+81-466-29-4412 e-mail: yoshikazu.ohtacom
Corresponding author: Yoshikazu Ohta, PhD. Address: Takeda Pharmaceutical Company Ltd, Fujisawa, Kanagawa 251-8555 Japan. Tel:+81-466-32-2621 Fax:+81-466-29-4412 e-mail: yoshikazu.ohtacom