Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2013; 4(6):447-457. doi:10.7150/jca.6896 This issue Cite
Research Paper
Wnt and the Cancer Niche: Paracrine Interactions with Gastrointestinal Cancer Cells Undergoing Asymmetric Cell Division
Hong-Wu Xin1, Chenwi M. Ambe1, Satyajit Ray1, Bo-Kyu Kim1, Tomotake Koizumi1, Gordon W. Wiegand1, Danielle Hari1, John E. Mullinax1, Kshama R. Jaiswal1, Susan H. Garfield2, Alexander Stojadinovic3,4, Udo Rudloff1, Snorri S. Thorgeirsson2, ![]() , Itzhak Avital1,4,5,
, Itzhak Avital1,4,5, ![]()
1. Surgery Branch, Center for Cancer Research, National Cancer Institute, National Institutes of Health, Bethesda, MD 20892, USA;
2. Laboratory for Experimental Carcinogenesis, Center for Cancer Research, National Cancer Institute, National Institutes of Health, Bethesda, MD 20892, USA;
3. Department of Surgery, Division of Surgical Oncology, Walter Reed National Military Medical Center, USA,
4. Department of Surgery, Uniformed Services University of the Health Sciences, Bethesda, MD, USA;
5. Bon Secours Cancer Institute, Richmond, VA 23230, USA.
Received 2013-6-12; Accepted 2013-6-30; Published 2013-7-2
Abstract
Objective: Stem-like cancer cells contribute to cancer initiation and maintenance. Stem cells can self-renew by asymmetric cell division (ACD). ACD with non-random chromosomal cosegregation (ACD-NRCC) is one possible self-renewal mechanism. There is a paucity of evidence supporting ACD-NRCC in human cancer. Our aim was to investigate ACD-NRCC and its potential interactions with the cancer niche (microenvironment) in gastrointestinal cancers.
Design: We used DNA double and single labeling approaches with FACS to isolate live cells undergoing ACD-NRCC.
Results: Gastrointestinal cancers contain rare subpopulations of cells capable of ACD-NRCC. ACD-NRCC was detected preferentially in subpopulations of cells previously suggested to be stem-like/tumor-initiating cancer cells. ACD-NRCC was independent of cell-to-cell contact, and was regulated by the cancer niche in a heat-sensitive paracrine fashion. Wnt pathway genes and proteins are differentially expressed in cells undergoing ACD-NRCC vs. symmetric cell division. Blocking the Wnt pathway with IWP2 (WNT antagonist) or siRNA-TCF4 resulted in suppression of ACD-NRCC. However, using a Wnt-agonist did not increase the relative proportion of cells undergoing ACD-NRCC.
Conclusion: Gastrointestinal cancers contain subpopulations of cells capable of ACD-NRCC. Here we show for the first time that ACD-NRCC can be regulated by the Wnt pathway, and by the cancer niche in a paracrine fashion. However, whether ACD-NRCC is exclusively associated with stem-like cancer cells remains to be determined. Further study of these findings might generate novel insights into stem cell and cancer biology. Targeting the mechanism of ACD-NRCC might engender novel approaches for cancer therapy.
Keywords: Cancer Stem Cells, Asymmetric Cell Division, Non-Random Chromosomal Cosegregation, Microenvironment.
INTRODUCTION
Stem cells can self-renew by symmetric cell division (SCD) or asymmetric cell division (ACD) where each daughter cell might assume different fates. Asymmetric cell division with non-random chromosomal cosegregation (ACD-NRCC) is a putative mechanism of cell self-renewal. However, ACD-NRCC is not necessarily indicative of self-renewal or asymmetric fate of daughter cells.
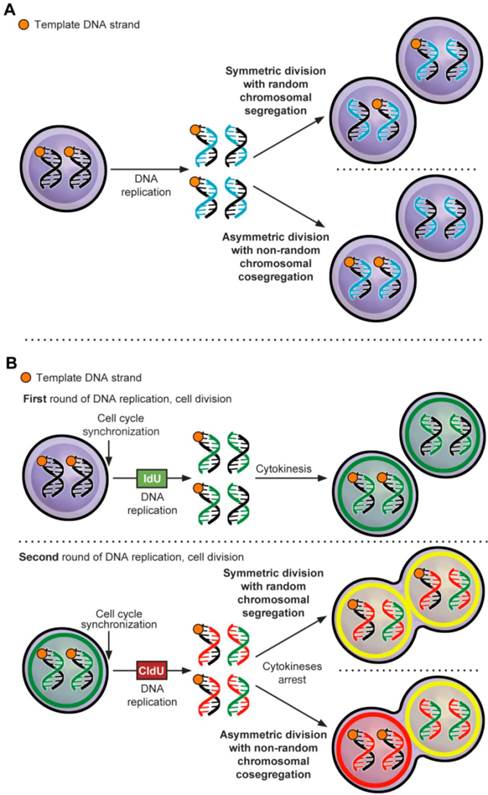
ACD-NRCC is defined as that each chromosome in a stem cell contains one template DNA strand that is conserved during ACD (figure 1A) [1]. By maintaining the template DNA strands within one daughter cell, stem cells can avoid accumulation of mutations stemming from DNA replication errors. Although, this principle has been demonstrated in various cells [2] some investigators were unable to confirm the existence of ACD-NRCC [3-8]. These inconsistent findings within the same cell types have provoked controversy, and have suggested a contributory role of the cancer niche/microenvironment [1-5, 7-12]. It's known that niche plays an important role in regulating stem cell ACD via Wnt signaling [13], however a role of niche via Wnt signaling has not been reported in regulating ACD-NRCC. Recently, we have demonstrated ACD-NRCC in real time, in live cells using real time confocal cinematography [14].
Asymmetric cell division via non-random chromosomal cosegregation (ACD-NRCC). (A) ACD-NRCC is proposed as one potential mechanism by which stem cells self-renew. It is hypothesized that stem cells contain template DNA strands that are conserved during asymmetric cell divisions (orange). By segregating the “template DNA strands” into daughter cells destined to become stem cells, stem cells could avoid propagation of DNA replication errors. This is a potential mechanism by which mutations are preferentially segregated into daughter cells destined to differentiate and are eventually eliminated. (B) Double labeling procedure for the detection of ACD-NRCC (Additional file 1: figure S1 and Materials and Methods; IdU (Green): Iodo-deoxyuridine; CldU (Red): Chloro-deoxyuridine).
Gastrointestinal cancers may develop in tissues harboring cells that undergo ACD-NRCC [1, 15, 16]. Previously, detection of ACD-NRCC relied exclusively on fixed cells, which hindered correlative gene expression and functional studies [2, 9]. Instead, we chose to isolate live cells undergoing ACD-NRCC and study this unique cell division process in gastrointestinal cancer cells, and investigate their potential interactions with the cancer niche. Elucidating the mechanisms of ACD-NRCC (and potential self-renewal) in cancer cells may provide novel insights into cancer maintenance and establish the basis for novel therapeutic approaches, which may target ACD-NRCC.
MATERIALS AND METHODS
Cells
Cells and culture conditions used in this study are detailed in Additional file 1: table S1.
DNA double-labeling-procedure
DNA double-labeling technique was used to detect ACD-NRCC (figure 1B, Materials and Methods, Additional file 1: table S1, S2 and S3) [9, 17, 18]. DNA double labeling was performed with minor modifications, as previously described by Rando et al. (figure 1B, Additional file 1: figure S1A, and Supplementary methods) [9]. To observe cells arrested during mitosis, cells were plated singly with and without 2μM Cytochalasin D (Sigma, Additional file 1: figure S1B and table S3). Double labeling was done with and without Wnt antagonist (10 μM of IWP2, Stemgent). siRNA-TCF4 (ON-TARGETplus, Dharmacon) or control siRNA (Dharmacon) were transfected using Lipofectamine-2000 according to manufacturer instruction (Invitrogen, Supplementary methods).
Immunofluorescence staining
Immunofluorescence staining was performed as previously described by Rando TA et al. with minor modifications (Supplementary methods) [9].
Fluorescence confocal microscopy
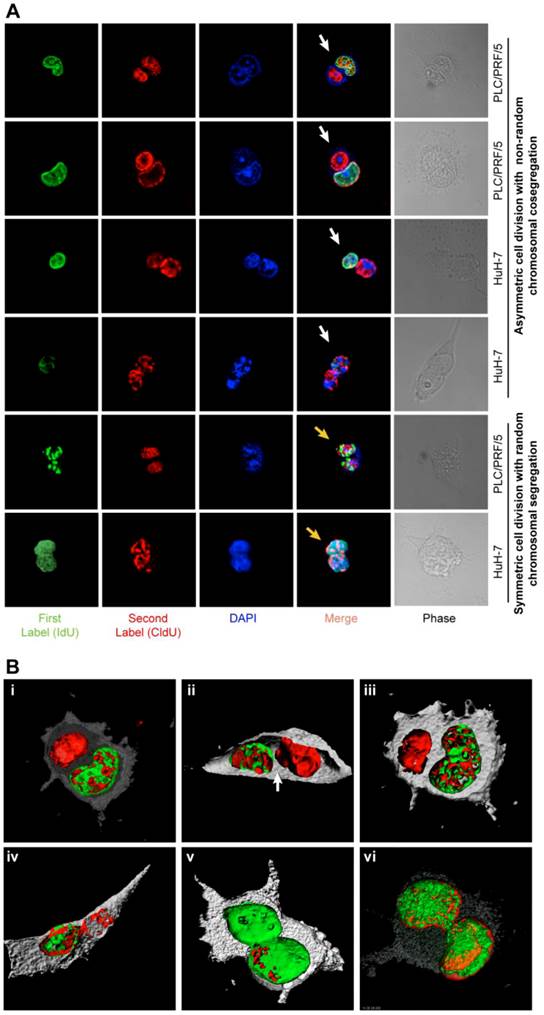
In order to accurately detect cells arrested in cytokinesis, we scored only isolated couplets, 100-130 couplets per condition, in triplicates (n=3 to 15). Confocal images and Z-stacks were generated with Bitplane's (Zurich, Switzerland) and Imaris software (v6.0). To clearly define the positions of the nuclei, 3D rendering images were obtained (figure 2, Additional file 1: figure S2, Supplementary methods). TCF4 staining-intensity was measured using LSM ImageBrowser 4.0 (Zeiss).
ACD-NRCC and SCD is shown in human liver cancer cells. (A) Fluorescent microscopy images showing ACD-NRCC (white arrow) and SCD with random chromosomal segregation (yellow arrow) in human liver cancers. The top four rows shows ACD-NRCC: Green fluorescent IdU is seen only in one nucleus after two cell cycles, while red fluorescent CldU is seen in both nuclei within the same cell arrested in cytokinesis. The bottom two rows show SCD, where both nuclei within one cell arrested in cytokinesis incorporated both nucleotides (Additional file 1: figure S2). (B) Three dimensional confocal microscopy images showing human liver cancer cells and lung cancer (iv) undergoing ACD-NRCC. In (i), (ii) and (iii), we show two nuclei in the same cytoplasmic space without intervening cytoplasmic membrane during ACD-NRCC (white arrow; Additional file 1: figure S2 and Additional file 2: Movie S1). In (v) we show ACD-NRCC after reverse labeling. In (vi) we show SCD for comparison.
Isolation of live cells undergoing ACD-NRCC and live label retaining cancer cells (LRCC)
Live cells undergoing ACD-NRCC and live LRCC were isolated as previously described in Hari et al., and Xin et al. (Additional file 1: figure S3A) [14, 19].
Cell viability and toxicity assays
Cell viability was measured using kit-8 (Dojindo, Japan) and cell toxicity using the ApoTox-Glo assay kit (Promega, Madison, WI) according to the manufacturer protocol (Supplementary Methods).
Isolation and analysis of the side population
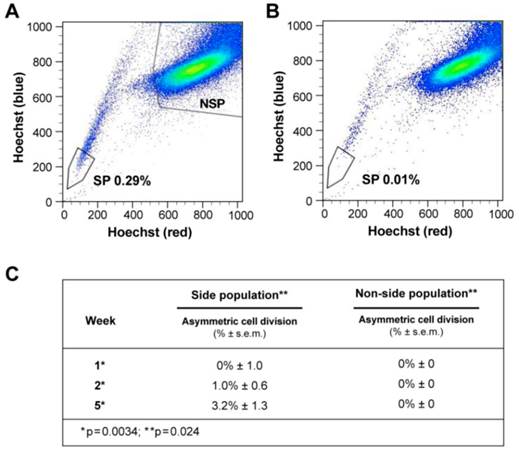
Side population (SP) cells were isolated as previously described (Supplementary methods, figure 3A-B) [20]. SP cancer cells are thought to represent putative tumor-initiating/cancer-stem-cells (figure 3A-B, Additional file 1: table S8) [21]. SP and non-SP cells of liver cancer cells were plated initially at very low concentration and allowed to proliferate.
ACD is detected preferentially in the Side Population of liver cancer cells. (A-B) FLOW CYTOMETRY sorting images of side population (SP) and non-side population cells (NSP). The side population is based on the ability of the ABCG2 transporter to efflux Hoechst 33342 (Ho). (B) In order to identify the population of cells that efflux Ho specifically by the ABCG2 transporter, we used Verapamil to block the activity of the ABCG2 transporter. (A) Here we show that the SP comprises 0.28% of the total cell population of Huh-7 liver cancer cells. (C) Side-population (SP) and non-SP hepatocellular carcinoma cells were plated initially at low concentrations, then allowed to proliferate, followed for 5 weeks, and tested for the presence of ACD-NRCC. At one week, SP cells did not exhibit ACD-NRCC, but when left to proliferate and differentiate while generating non-SP cells, demonstrated increasing levels of ACD-NRCC (p=0.0034); the non-SP cells neither generated SP cells nor demonstrated ACD-NRCC (p=0.024). While ACD-NRCC was never detected in NSP or CD133-negative cells (Vide Infra, figure 4), the maximal detection rate in total cells, in SP cells or in CD133+ cells seem to be constant suggesting that per a given condition the rate of ACD-NRCC is constant (steady state rate).
CD133+ cell isolation
CD133+ and CD133- cells were isolated and enriched over a second column by magnetic-activated cell sorting (MACS), using the indirect CD133 MicroBead kit (Miltenyi Biotec Inc., Auburn, CA, USA) according to manufacturer's protocol (Supplementary methods).
Conditioned media
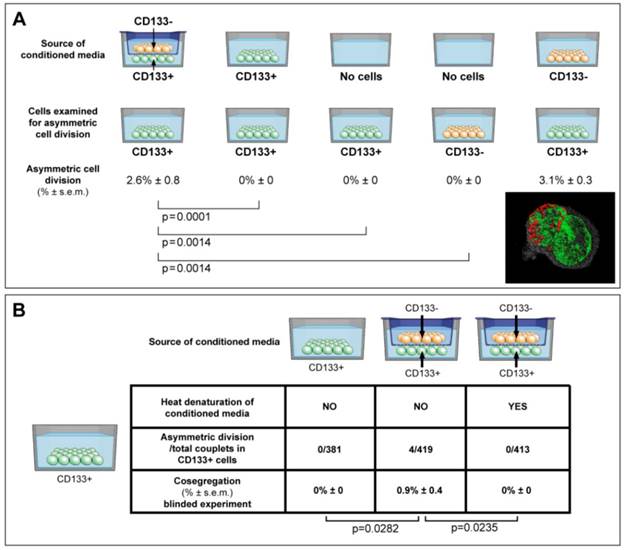
Conditioned media were collected aseptically, filtered through 0.22 µm filter units, and mixed with normal growth media at a ratio of 1:3 (figure 4A, supplementary methods). Denatured conditioned media from CD133-/CD133+ dual chambers was boiled for 5 minutes, and 5% FBS was added to a total protein concentration equal to normal growth media. Specific gravity, pH and protein concentration were determined with Specific Gravity Bottle (Crystalgen, Inc., USA), pH Meter (HANANA instruments, USA), and Spectrophotometer (Thermo Fisher Scientific, USA).
ACD is detected preferentially in CD133+ cells, and is modulated by the cancer microenvironment in a paracrine nature. (A) Here we show that ACD-NRCC is detected in CD133+ cells of Huh-7 liver cancer cells; no ACD-NRCC was detected in CD133-negative cells under any-condition. Additionally, we show that in order for CD133+ cells to undergo ACD-NRCC they must be cultured together with CD133-negative cells. The effect of CD133-negative cells on CD133+ cells is paracrine in nature. Thus, the effect of CD133-negative cells on CD133+ cells undergoing ACD-NRCC is not dependent on cell-to-cell contact. CD133-negative or CD133+ cells growing separately alone do not undergo ACD-NRCC. All experiments were repeated three times in a prospective blinded fashion. (B) Here we show that the permissive effect of CD133-negative cells on CD133+ cells undergoing ACD-NRCC is heat sensitive, and can be abolished by heat denaturation. While ACD-NRCC was never detected in CD133-negative cells or NSP cells (figure 3), the maximal detection rate in total cells, in CD133+ cells or in SP cells seem to be constant suggesting that per a given condition the rate of ACD-NRCC is constant (steady state rate).
Gene expression analysis
Total RNA was isolated using miRNeasy Mini kit and RNase-Free DNase Set (QIAGEN) following the manufacturer's protocol. All reagents for genomic DNA elimination, reverse-transcription, pre-amplification, and real-time qRT-PCR experiments for Human Stem Cell Pathway, Wnt and Pluripotency Pathway Arrays were done following the manufacturer's protocol (SABiosciences, Frederick, MD). Primers for individual genes: TCF4, TCF7, SOX17 and CSNK2A1 were purchased from Qiagen. We used the Ingenuity Pathway software for pathway analysis (IPA 9.0, supplementary methods).
Statistics
For full discussion see supplementary statistics. In brief, for detecting any ACD-NRCC, we used the exact binomial test with a null hypothesis of 0.00001. (B) To test for significance of the relative proportions of cells undergoing ACD-NRCC between tested groups, we used the Poisson method (figure 3C). (C) For the observed effect of the niche on ACD via non-random chromosomal cosegregation (figure 4A), we used the Fisher's exact test. Statistical significance was defined as p value <0.05.
RESULTS
Subpopulations of gastrointestinal cancer cells undergo ACD-NRCC
Symmetrically dividing cells incorporated both nucleotides (IdU and CldU) into the nuclei of both daughter cells. Cells undergoing potential ACD-NRCC incorporated both nucleotides into only one of the daughter cells' nuclei while the other nucleus incorporated only one nucleotide (figure 2A-B, Additional file 2: movie S1 and Additional file 1: figure S2). Reverse labeling resulted in a similar pattern (figure 2B-v, Additional file 1: figure S2B and table S4). We validated these results in 7 different cell lines and fresh surgical specimens using this DNA double labeling method; ACD-NRCC was observed in 1.5% to 6.3% (n=21) of cells tested (Additional file 1: table S5). To identify asymmetrically dividing cells, we used confocal microscopy, and confirmatory Z-stacking with three-dimensional-rendering of cells arrested in cytokinesis-mitosis (figure 1B, 2, and Additional file 1: figure S2). We further validated ACD-NRCC by isolating live cells undergoing ACD-NRCC using flow-cytometry (1.6% ± 0.3%, n=17; Additional file 1: figure S3B-C, Materials and Methods). Cell viability and toxicity were tested with and without labeled-nucleotides. No statistically significant differences were evident between cells undergoing ACD-NRCC vs. SCD (Additional file 1: figure S3D-E).
ACD-NRCC is a statistically significant phenomenon, occurring in 1 to 6% of 14 cancer cell lines (p<0.0001, n=63, supplementary-statistics), and in non-malignant liver cells (THLE-2 and THLE-3: 1.3% ± 0.3%, and 2.6 ± 0.6%, respectively) as demonstrated in three different methods (Additional file 1: table S5-to-S7). In addition to the methods above, these results were validated using two additional FACS-based-methods: (1) double labeling FACS-based method to isolate 25,630 of 1,026,314 cells undergoing ACD-NRCC (Additional file 1: figure S3, table S6); (2) single labeling FACS-based method to isolate live cells undergoing ACD-NRCC (Additional file 1: table S7, Materials and Methods). The single labeling method yielded over 5 million cells undergoing ACD-NRCC derived from 11 different cell lines and cells from fresh surgical specimens.
Side-Population (SP) cancer cells undergo ACD-NRCC
At one week, SP cells generated non-SP cells while non-SP cells generated only more non-SP cells (figure 3C, supplementary statistics) [20]. At one, two and five weeks SP cells exhibited 0%, 1%, and 3.2% ACD-NRCC, respectively (p<0.0034, n=3). Non-SP cells did not undergo ACD-NRCC, during the same time periods (P=0.024). To validate these findings, we isolated SP and non-SP cells from four different gastrointestinal cancers and melanoma. SP and non-SP cells were cultured for three weeks and tested for ACD-NRCC. In a blinded experiment, we detected ACD-NRCC only in SP cells (1.8% ± 0.33, p=0.004, n=12). OV6 is a marker associated with liver progenitors; it was found to segregate with the template DNA strand in SP cells undergoing ACD-NRCC; it was not detected in non-SP cells (Additional file 1: figure S4) [22].
The cancer niche interacts with CD133+ cells undergoing ACD in a paracrine fashion
CD133+ cells are thought to represent another class of putative tumor-initiating/cancer-stem-cells [23]. We detected ACD-NRCC in whole liver cancer cell-lines (1.0% ± 0.05, n=3). The absence of ACD-NRCC in either CD133+ or CD133-negative cells alone, prompted us to test for potential interactions between CD133+ and CD133-negative cells, using a dual chamber culturing system with semi-permeable membrane. We detected ACD-NRCC when CD133+ cells were in direct contact with CD133-negative cells (1.3% ± 1.0, n=3). Using the dual-chamber culture system, ACD-NRCC was detected in CD133+ cells (2.0% ± 1.0, n=6) that were exposed indirectly to CD133-negtive cells (figure 4A). Conversely, no ACD-NRCC was detected in CD133-negative cells indirectly exposed to CD133+ cells (Supplementary Materials and Methods). These results support our previous data investigating SP and non-SP cells, suggesting that ACD-NRCC is preferentially detected in SP and CD133+ cells.
Consequently we hypothesized that the cancer niche, i.e. the non-stem-cancer-cell population (CD133-negative cells), interacts indirectly with CD133+ cells undergoing ACD-NRCC. Therefore, we tested the effect of various conditioned media on ACD-NRCC (figure 4A). These experiments were conducted with the scientist testing for ACD-NRCC blinded to the conditions being tested (n=6). The physical properties of the conditioned media and the fresh media (pH, protein concentration and specific gravity) were not significantly different (P>0.05). Conditioned media from dual-chambers (containing CD133+ and CD133-negative cells), and from CD133-negative cells alone induced CD133+ cells to undergo ACD-NRCC (2.6% ± 0.8, and 3.1% ± 0.3, respectively). ACD-NRCC was not detected in CD133+ cells cultured in conditioned media from CD133+ cells (p=0.0001) or in CD133+ cells cultured alone (p=0.014), in contrast to conditioned media from dual-chambers. CD133-negative cells (cancer niche cells) did not undergo ACD-NRCC (p=0.014).
These results were further validated in blinded experiments aimed at determining whether heat-denaturation of the conditioned media would abrogate ACD-NRCC (figure 4B). ACD-NRCC was neither detected in CD133+ cells grown in heat-denatured conditioned media from dual chambers (p=0.023), nor in CD133+ cells alone (negative control). ACD-NRCC was detected in CD133+ cells cultured in media from dual chambers without heat-denaturation (p=0.028, positive control).
Taken together our data suggest that ACD-NRCC is preferentially detected in SP and CD133+ cells, and that the cancer niche/microenvironment can interact with cells undergoing ACD-NRCC in a heat-sensitive paracrine fashion.
The WNT pathway's transcription factor TCF is asymmetrically localized in cells undergoing ACD
The Wnt pathway plays an important role in stem cells biology, self-renewal, and stem cells pluripotency [24, 25]. Using similar method for the detection of ACD-NRCC (Materials and Methods), we tested 6 key Wnt proteins from each of the 3 cellular compartments (cell membrane, cytoplasm and nucleus) in liver cancer cells undergoing mitosis. Only TCF4 was distributed asymmetrically (3.1% ± 1.5; 19/619 mitoses, n=3), while Frizzled7, LRP5, Axin2, APC and β-catenin were distributed symmetrically in all cells tested (figure 5A-B, Materials and Methods). To further validate these results, we tested TCF4 gene expression in isolated cells undergoing ACD-NRCC, and cells undergoing SCD (Additional file 1: figure S3). Using real-time qRT-PCR, TCF4 was up regulated in cells undergoing ACD-NRCC when compared to cells undergoing SCD in all cell lines tested (3.8 ± 0.4 fold, p< 0.00003, figure 5C).
Wnt pathway genes are expressed asymmetrically in cells undergoing asymmetric cell division (ACD-NRCC). We studied representative Wnt associated proteins representing each of the cellular compartments (membrane, cytoplasm and nucleus). TCF4 was the only Wnt associated protein distributed asymmetrically during liver cancer cell divisions (3.1% ± 1.5%). (A) Using confocal microscopy with tridimensional rendering, we show asymmetric distribution of TCF4, and (Green=TCF4, Blue=DAPI and Red=CFSE), and (B) symmetric distribution of TCF4. (C) We isolated live cells undergoing ACD-NRCC or SCD; using qRT-PCR, we show that TCF4 is upregulated in cells undergoing ACD-NRCC when compared to cells undergoing SCD. (D) We performed Wnt SuperArray analysis on cells undergoing ACD-NRCC or SCD. Among 84 Wnt associated genes tested only 5 were found to be differentially expressed, TCF4, TCF7, CSBK2A1 and Sox17 were upregulated, and RB1 was down-regulated by cells undergoing ACD-NRCC.
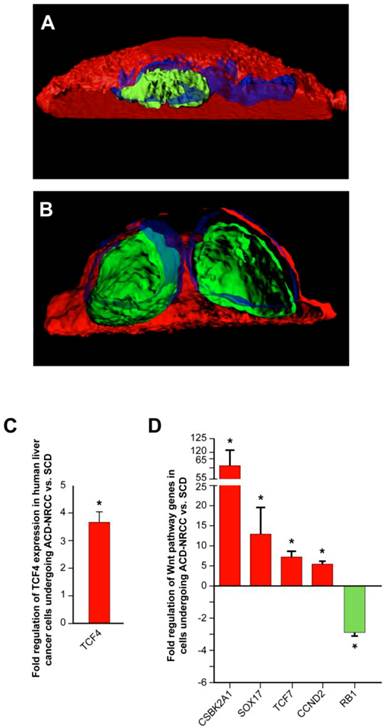
To further test potential interactions between the Wnt pathway and ACD-NRCC, we performed comprehensive Wnt gene expression analysis on liver cancer cells undergoing SCD vs. ACD-NRCC (Materials and Methods). Of 84 tested Wnt genes, only 5 were differentially expressed in cells undergoing ACD-NRCC: CSNK2A1, which activates the Wnt pathway (61.7 ± 9.1 fold, p=0.024), TCF4 (3.8 ± 0.4 fold, p< 0.00003), TCF7 (13.1 ± 6.5 fold, p=0.038), Sox17 (7.4 ± 1.3 fold, p=0.0082), and RB1 (-3.86 ± 0.28 fold, p<0.011; figure 5D). These data were validated using qRT-PCR for individual genes (Additional file 1: figure S5). Using the ingenuity pathway analysis tool, and stem cells gene expression analysis (Additional file 1: figure S6), we propose a pathway map for cells undergoing ACD-NRCC (Additional file 1: figure S7).
Inhibition of Wnt suppresses ACD-NRCC and the asymmetric expression of Wnt genes
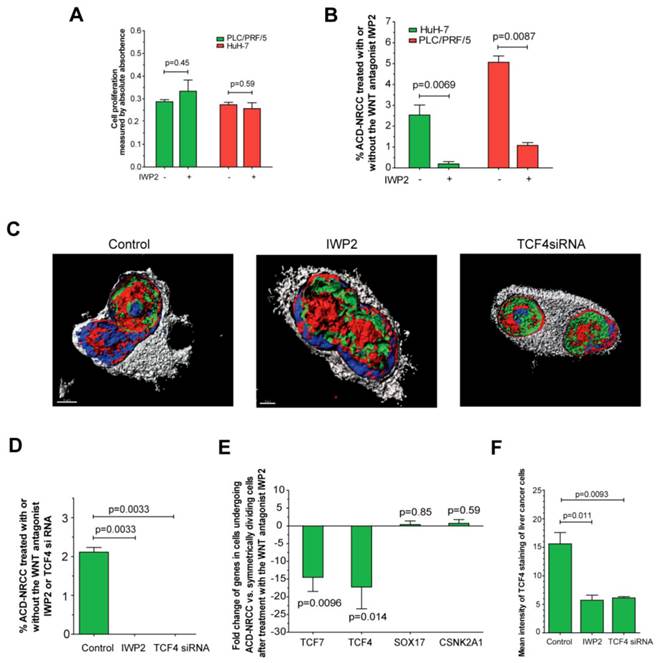
We found that several Wnt pathway genes are differentially expressed in cells undergoing ACD-NRCC vs. SCD. Therefore, we tested the effects of Wnt activation and inhibition on ACD-NRCC (Materials and Methods). The rate of ACD-NRCC was unaffected by Wnt activation with Wnt3 (Wnt agonist). We used the Wnt3 agonist because we found that Wnt3-receptor is expressed equally on cells undergoing ACD-NRCC or SCD. Because we did not detect changes in the rate of ACD-NRCC after activation of the Wnt pathway via Wnt3 receptor, we next tested the effects of Wnt inhibition on ACD-NRCC. The effects of IWP2 (Wnt-antagonist) and siRNA-TCF4 on ACD-NRCC were then tested. Without affecting cell proliferation (figure 6A) a 5-to-12 fold reduction in the rate of ACD-NRCC was evident in live cells after treatment with IWP2 (5.1% ± 0.3% vs. 1.1% ± 0.1%, p=0.0087 for PLC/PRF/5; and, 2.5% ± 0.5% vs. 0.2% ± 0.1%, p=0.0069 for HuH-7; figure 6B). The ability to detect ACD-NRCC and asymmetric distribution of TCF4 protein in fixed cells was abolished by siRNA-TCF4 (2.1% ± 0.1% vs. 0, p=0.0033; figure 6C-D). IWP2 reversed or reduced the expression pattern of TCF4 (-17.2 ± 6.1 fold, p=0.0096), TCF7 (-14.5 ± 4.0 fold, p=0.014), Sox17 (0.4% ± 0.9 fold, p=0.85) and CSNK2A1 (0.7 ± 1.1% fold, p=0.59; figure 5, 6E and S5). IWP2 and siRNA-TCF4 reduced the expression of TCF4 protein by 63% (15.6 ± 2.0 vs. 5.7 ± 0.9, p=0.011) and 61% (15.6 ± 2.0 vs. 6.1 ± 0.2, p=0.0093; figure 6F, Additional file 1: figure S8). These results implicate the Wnt pathway in ACD-NRCC.
Inhibition of Wnt results in significant suppression of ACD-NRCC. We detected ACD-NRCC or SCD before and after treatment with the Wnt-antagonist IWP2 or siRNA-TCF4. (A) IWP-2 didn't affect cell proliferation (0.29 ± 0.01 vs. 0.33 ± 0.0, p=0.45 for PLC/PRF/5; 0.27 ± 0.01% vs. 0.25 ± 0.03, p=0.59 for HuH-7). (B) To understand Wnt effects on ACD-NRCC in a quantitative manner, we tested for ACD-NRCC in live cells before and after treatment with the Wnt-antagonist IWP2. The suppression of ACD-NRCC after treatment with the Wnt-antagonist IWP2 was statistically significant. (C-D) To further validate these results and because TCF4 was differentially expressed in cells undergoing ACD-NRCC, we tested for ACD-NRCC by confocal microscopy in fixed cells before and after treatment with the Wnt-antagonist IWP2 or TCF4-siRNA. We detected ACD-NRCC in cells before treatment with IWP2 or TCF4-siRNA. After treatment with the Wnt-antagonist IWP2 or knock down of TCF4, we couldn't detect cells undergoing ACD-NRCC; only cells undergoing SCD were observed. (E) Treatment with the Wnt-antagonist IWP2 reversed or reduced the differential expression of TCF4, TCF7, SOX17 and CSNK2A1 in cells undergoing ACD-NRCC vs. SCD (figure 5 and S5). (F) Immunofluorescence staining for TCF4 before and after treatment with IWP2 or TCF4-siRNA showed that the Wnt antagonist IWP-2 or TCF4-siRNA reduced TCF4 levels by 64% (p=0.011) and 61% (p=0.0093), respectively (Additional file 1: figure S8).
DISCUSSION
Albeit controversial, recent evidence suggests the existence of stem-like tumor initiating cancer cells. ACD-NRCC is one potential way by which stem cells self-renew. Here we show that ACD-NRCC was detected preferentially in SP and CD133+ cells. ACD-NRCC occurred independent of cell-to-cell contact; instead, the microenvironment seemingly induced CD133+ cells to undergo ACD-NRCC in a heat-sensitive paracrine fashion. Mechanistically, we show that cells undergoing ACD-NRCC segregate asymmetrically the Wnt transcription factor TCF, and other Wnt genes. ACD-NRCC was unaffected by agonist-dependent Wnt pathway activation; however, inhibition of Wnt and knock down of TCF resulted in a significant suppression of ACD-NRCC.
Pine et al reported detection of ACD-NRCC in fixed lung cancer cells [11]. Here we show ACD-NRCC in several fixed and extend the findings to live gastrointestinal cancer cells. We validated these observations using two methods: DNA double and single labeling (label retention) [9, 17, 18]. The rate of ACD-NRCC detection was comparable between the two methods. Once thought to be a rare phenomenon, it is possible that ACD-NRCC occurs more commonly, and amongst cancers of diverse origins.
Observing this phenomenon in various GI cancers, we hypothesized that defects in ACD-NRCC could be an etiological factor in malignancy. Such a mechanism, potentially involving linking template DNA strands from multiple chromosomes to “one-side” of the mitotic plane, would be prone to “breaks” leading to aneuploidy and eventually cancer. Defects in asymmetric cell division in the drosophila results in cancer [26]. Defects in ACD-NRCC provide another explanation for how tissue stem cells initiate cancer.
A potential source of bias in our study is the use of labeled DNA nucleotides. It is conceivable that labeled-nucleotides could affect proliferation, cell cycle, cell survival, DNA replication, and cell division. However, reported data is scant, and contradictory. To test this potential source of bias, we tested cell viability/proliferation/toxicity in labeled and non-labeled cells, and found no difference in viability, proliferation and/or toxicity (Additional file 1: figure S3D-E). Based on these data, our findings do not appear to be due to artifact (Supplementary Discussion).
We showed that ACD-NRCC is detected both in cancer cells previously described as tumor-initiating cells or putative stem-like cancer cells (Side Population and CD133+ cells), and associated with OV6 expression (A marker associated with liver progenitors). Others showed that cells undergoing ACD-NRCC express stem cells markers [9, 11], and that ACD-NRCC is not associated with differentiation markers [11]. Additionally, asymmetric cell division was detected in intestinal stem cells [27, 28], and after tissue injury [26]. However, based on our data we cannot associate ACD-NRCC specifically or directly with cancer-stem cells or even a different cell fate [10].
Based on this work, we can't attribute ACD-NRCC exclusively to cancer stem cells. However, recently we published a paper titled 'Tumor-initiating label retaining cancer cells in human gastrointestinal cancers undergo asymmetric cell division' [14]; in that work, we describe how some cancer cells undergoing ACD-NRCC exhibit exquisite tumor initiating capacity and multipotency gene expression profile. Moreover, there are significant differences between cells undergoing ACD-NRCC and cells undergoing SCD in terms of stem-cells and pluripotency gene expression profiles. The former exhibit stem cells and pluripotency gene expression profile with certain genes like Sox2 being expressed several hundred fold higher. In addition, we reported that the label retaining liver cancer cells are relatively resistant to the targeted cancer drug sorafenib [29]. In general, we hold the opinion that a stem-like-cancer cell is a more appropriate term. Since we can't associate directly cancer stem cells or stem-like-cancer cells with ACD-NRCC based purely on the current paper and our previous work was just recently published and was not yet validated by an independent group, we prefer to use the term "putative stem-like-cells" in the body of the text.
We found that the cancer niche can induce CD133+ cells to undergo ACD-NRCC in a paracrine fashion that does not require cell-to-cell contact. This effect could be abolished by heat denaturation of the conditioned media, suggesting a potential soluble factor. In contrast, Pine et al. found that lung CD133+ cells undergo ACD-NRCC when in direct contact with CD133- cells. One potential explanation could be that we used DNA-double-labeling and Pine et al. used single labeling (label retention). Another potential explanation could be that various cancers (lung vs. GI cancers) respond differently to environmental signals. These observations and the potential effects of the cancer niche (microenvironment) on ACD-NRCC carry important implications to cancer and stem cells biology.
The molecular mechanism of ACD-NRCC is poorly understood. This is the first study to investigate gene expression in live cells undergoing ACD-NRCC. Cells undergoing ACD-NRCC differentially distribute TCF4 (Wnt transcription factor), up-regulate Wnt (TCF4, TCF7, CSNK2A1, and CCND2, and Sox17) and pluripotency/stem-cell (Sox2, GDF3, UTF1 and NEUROG2) genes, suggesting a stem-cell-like gene expression profile [23]. Using Ingenuity Pathway Analysis, we propose a Wnt-pluripotency pathway map for ACD-NRCC (Additional file 1: figure S7). Most of the up-regulated genes in cells undergoing ACD-NRCC found in this study were previously reported to be oncogenes, while down-regulated genes are tumor suppressor genes [26]. Furthermore, from a mechanistic point of view, we describe the potential role that the Wnt pathway plays in ACD-NRCC. Although a Wnt agonist didn't stimulate ACD-NRCC, exposure to Wnt antagonist IWP2 and siRNA-TCF4 resulted in significant suppression of ACD-NRCC, and asymmetric distribution of TCF4. Here we show that Wnt is involved in ACD-NRCC but its precise role in the complex interactions within cells undergoing ACD-NRCC, demand further elucidation by independent groups.
Taken together, we show that subpopulations of gastrointestinal cancer cells can undergo ACD-NRCC. ACD-NRCC was detected preferentially in SP and CD133-positive cells, and was independent of cell-to-cell contact. The cancer niche, interact in a paracrine fashion, with cells undergoing ACD-NRCC, which is suppressed by Wnt antagonist and siRNA-TCF. Targeting the mechanism of ACD-NRCC might result in novel approaches for cancer therapy.
Supplementary Material
Additional File 1Supplementary Tables, Supplementary Figures, Supplementary Discussion and Supplementary Methods.
Supplementary Movie
Abbreviations
ACD-NRCC: Asymmetric Cell Division with non-Random Chromosomal Cosegregation
SCD: Symmetric Cell Division
ACD: Asymmetric Cell Division
MACS: Magnetic Activated Cell Sorting
SP: Side population
NSP: Non-Side Population
Acknowledgements
We thank Langston Lim and Poonam Annan for their excellent help in confocal microscopy.
Funding
NCI grant 1ZIABC011005.
Patient consent
Obtained.
Ethics approval
Approved by NIH.
Contributors
HWX, SST and IA conceived and designed the experiments; HWX, CA, SR, BKK, TK, GWW, DH and JEM performed the experiments; HWX, SHG, SST, IA analyzed the data; HWX, CA, KRJ, AS, UR, SST and IA wrote the paper.
Competing interest
The authors indicate no potential conflicts of interest.
References
1. Cairns J. Mutation selection and the natural history of cancer. Nature. 1975;255:197-200
2. Rando TA. The immortal strand hypothesis: segregation and reconstruction. Cell. 2007;129:1239-43
3. Kiel MJ, He S, Ashkenazi R, Gentry SN, Teta M, Kushner JA. et al. Haematopoietic stem cells do not asymmetrically segregate chromosomes or retain BrdU. Nature. 2007;449:238-42
4. Fei JF, Huttner WB. Nonselective sister chromatid segregation in mouse embryonic neocortical precursor cells. Cereb Cortex. 2009;19(Suppl 1):i49-54
5. Kuroki T, Murakami Y. Random segregation of DNA strands in epidermal basal cells. Jpn J Cancer Res. 1989;80:637-42
6. Barker N, van Es JH, Kuipers J, Kujala P, van den Born M, Cozijnsen M. et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature. 2007;449:1003-7
7. Sotiropoulou PA, Candi A, Blanpain C. The majority of multipotent epidermal stem cells do not protect their genome by asymmetrical chromosome segregation. Stem Cells. 2008;26:2964-73
8. Waghmare SK, Bansal R, Lee J, Zhang YV, McDermitt DJ, Tumbar T. Quantitative proliferation dynamics and random chromosome segregation of hair follicle stem cells. The EMBO journal. 2008;27:1309-20
9. Conboy MJ, Karasov AO, Rando TA. High incidence of non-random template strand segregation and asymmetric fate determination in dividing stem cells and their progeny. PLoS biology. 2007;5:e102. doi:06-PLBI-RA-2054R2 [pii]. 10.1371/journal.pbio.0050102
10. Lansdorp PM. Immortal strands? Give me a break. Cell. 2007;129:1244-7
11. Pine SR, Ryan BM, Varticovski L, Robles AI, Harris CC. Microenvironmental modulation of asymmetric cell division in human lung cancer cells. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:2195-200
12. Potten CS, Hume WJ, Reid P, Cairns J. The segregation of DNA in epithelial stem cells. Cell. 1978;15:899-906 doi:0092-8674(78)90274-X [pii]
13. Lu B, Jan LY, Jan YN. Asymmetric cell division: lessons from flies and worms. Current opinion in genetics & development. 1998;8:392-9
14. Xin HW, Hari DM, Mullinax JE, Ambe CM, Koizumi T, Ray S. et al. Tumor-initiating label-retaining cancer cells in human gastrointestinal cancers undergo asymmetric cell division. Stem Cells. 2012;30:591-8
15. Li F, Lu L, Lu J. Identification and location of label retaining cells in mouse liver. J Gastroenterol. 2010;45:113-21
16. Cotsarelis G, Sun TT, Lavker RM. Label-retaining cells reside in the bulge area of pilosebaceous unit: implications for follicular stem cells, hair cycle, and skin carcinogenesis. Cell. 1990;61:1329-37 doi:0092-8674(90)90696-C [pii]
17. Potten CS, Owen G, Booth D. Intestinal stem cells protect their genome by selective segregation of template DNA strands. Journal of cell science. 2002;115:2381-8
18. Smith GH. Label-retaining epithelial cells in mouse mammary gland divide asymmetrically and retain their template DNA strands. Development. 2005;132:681-7 doi:dev.01609 [pii]. 10.1242/dev.01609
19. Hari D, Xin HW, Jaiswal K, Wiegand G, Kim BK, Ambe C. et al. Isolation of live label-retaining cells and cells undergoing asymmetric cell division via nonrandom chromosomal cosegregation from human cancers. Stem cells and development. 2011;20:1649-58
20. Haraguchi N, Utsunomiya T, Inoue H, Tanaka F, Mimori K, Barnard GF. et al. Characterization of a Side Population of Cancer Cells from Human Gastrointestinal System. Stem Cells. 2006;24:506-13 doi:10.1634/stemcells.2005-0282
21. Ma S, Chan K-W, Hu L, Lee TK-W, Wo JY-H, Ng IO-L. et al. Identification and Characterization of Tumorigenic Liver Cancer Stem/Progenitor Cells. Gastroenterology. 2007;132:2542-56
22. Parent R, Marion MJ, Furio L, Trepo C, Petit MA. Origin and characterization of a human bipotent liver progenitor cell line. Gastroenterology. 2004;126:1147-56
23. Pera MF, Tam PP. Extrinsic regulation of pluripotent stem cells. Nature. 2010;465:713-20
24. Balciunaite G, Keller MP, Balciunaite E, Piali L, Zuklys S, Mathieu YD. et al. Wnt glycoproteins regulate the expression of FoxN1, the gene defective in nude mice. Nat Immunol. 2002;3:1102-8
25. Quyn AJ, Appleton PL, Carey FA, Steele RJ, Barker N, Clevers H. et al. Spindle orientation bias in gut epithelial stem cell compartments is lost in precancerous tissue. Cell stem cell. 2010;6:175-81
26. Wu PS, Egger B, Brand AH. Asymmetric stem cell division: lessons from Drosophila. Semin Cell Dev Biol. 2008;19:283-93 doi:S1084-9521(08)00009-8 [pii]. 10.1016/j.semcdb.2008.01.007
27. Lopez-Garcia C, Klein AM, Simons BD, Winton DJ. Intestinal stem cell replacement follows a pattern of neutral drift. Science. 2010;330:822-5
28. Snippert HJ, van der Flier LG, Sato T, van Es JH, van den Born M, Kroon-Veenboer C. et al. Intestinal crypt homeostasis results from neutral competition between symmetrically dividing Lgr5 stem cells. Cell. 2010;143:134-44
29. Xin HW, Ambe CM, Hari DM, Wiegand GW, Miller TC, Chen JQ. et al. Label-retaining liver cancer cells are relatively resistant to sorafenib. Gut. 2013 Feb 14. [Epub ahead of print]. PMID: 23411027
Author contact
![]() Corresponding author: Snorri S. Thorgeirsson, M.D., Ph.D. Head, Center of Excellence in Integrative Cancer Biology and Genomics Chief, Laboratory of Experimental Carcinogenesis, Center for Cancer Research, National Cancer Institute, NIH, 37 Convent Drive MSC 4262, Building 37, Room 4146A, Bethesda, MD 20892-4262, USA. Phone: 1+(301) 496-1450(O) Phone: 1+(301) 496-1935(W) Fax: 1+ (301) 496-0734 E-mail: snorri_thorgeirssongov Or Itzhak Avital, MD. Executive Medical Director, Bon Secours Cancer Institute, Richmond, VA 23230, USA. Email: itzhak.avitalcom.
Corresponding author: Snorri S. Thorgeirsson, M.D., Ph.D. Head, Center of Excellence in Integrative Cancer Biology and Genomics Chief, Laboratory of Experimental Carcinogenesis, Center for Cancer Research, National Cancer Institute, NIH, 37 Convent Drive MSC 4262, Building 37, Room 4146A, Bethesda, MD 20892-4262, USA. Phone: 1+(301) 496-1450(O) Phone: 1+(301) 496-1935(W) Fax: 1+ (301) 496-0734 E-mail: snorri_thorgeirssongov Or Itzhak Avital, MD. Executive Medical Director, Bon Secours Cancer Institute, Richmond, VA 23230, USA. Email: itzhak.avitalcom.