Journal of Cancer
ISSN: 1837-9664
3.2
Impact Factor
ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2013; 4(5):371-382. doi:10.7150/jca.6625 This issue Cite
Research Paper
Ex Vivo Derived Primary Melanoma Cells: Implications for Immunotherapeutic Vaccines
Robert Suriano 1, Shilpi Rajoria1, Andrea L.George1, Jan Geliebter1, Marc Wallack1, 2, Raj K. Tiwari1, ![]()
1. Department of Microbiology and Immunology, New York Medical College, Valhalla, New York, 10595;
2. Metropolitan Hospital Center GNS, Department of Surgery, 1901 1st Avenue, New York, NY 10029.
Received 2013-5-6; Accepted 2013-5-29; Published 2013-6-19
Citation:
Suriano R, Rajoria S, L.George A, Geliebter J, Wallack M, Tiwari RK. Ex Vivo Derived Primary Melanoma Cells: Implications for Immunotherapeutic Vaccines. J Cancer 2013; 4(5):371-382. doi:10.7150/jca.6625. https://www.jcancer.org/v04p0371.htm
Other stylesAbstract
Transformation of the pigment producing melanocytes into melanoma is a complex multi-step process involving the enhanced expression of various antigens considered as immunotherapeutic targets. Significant progress in melanoma research has been made over the years and has resulted in the identification of various antigens over expressed in melanoma as well as advances in immunotherapeutic treatments, which focus on modulating the immune systems response to melanoma. Despite these advances, incidences of melanoma are still on the rise thus warranting additional research in identifying new therapeutic treatments. Our focus is on developing a multivalent immunotherapeutic vaccine that targets various melanoma associated antigens. The approach focuses on the use of five primary patient derived melanoma cells (MEL-2, MEL-V, 3MM, KFM, and GLM-2, which have been characterized in this study. These cells express differential amounts of various melanoma associated antigens such as MART-1, gp100 (Pmel17), MAGE-A1 and tyrosinase as well a cell surface antigens essential for melanoma cell metastasis, such as CD146 and CD71. In addition these cells display differential in vitro migratory and invasive properties as well as have the ability to form solid tumors when implanted into BALB/c nude mice. The retention of the innate phenotype of these primary patient derived cells together with the expression of a multitude repertoire of melanoma associated antigens offers a novel opportunity to target melanoma so as to avoid immune evasion.
Keywords: immunotherapeutic vaccine, melanoma cells
Introduction
Melanoma, which is the most deadly form of skin cancer, arises from the pigment producing cells of the skin, known as melanocytes [1]. The National Cancer Institute has classified melanoma as the fifth most common occurring cancer in men and seventh most common occurring cancer in women. In addition, it is estimated by the American Cancer society that over 76,000 individuals will be diagnosed with melanoma and over 9,000 individuals will die from melanoma within the United States in 2013. To date, surgery remains the standard treatment for localized melanoma skin lesions and for metastatic melanoma, a single chemotherapeutic agent known as dacarbazine is used, which has a response rate of 10% to 15 % in patients and is not associated with long lasting responses as the median survival rate is approximately 9 months [2,3]. In essence, chemotherapy has lagged behind as an effective adjuvant thus warranting our investigation to alternative therapeutic modalities. Since melanoma is one of the most immunogenic tumors, with several melanoma associated antigens having been defined, it is important to develop a multivalent immunotherapeutic vaccine [4-9].
Immunotherapy of melanoma, which has been experimented with over the past 30 years and resulted in various clinical trials [4,10], has focused on four major areas to achieve maximal efficacy; immuno-activating cytokines, immuno-modulating antibodies, adoptive T-cell therapy, and vaccines. Interleukin -2 (IL-2) is one of two FDA approved compounds available to treat melanoma but is associated with high toxicity [11,5]. Within the past year, the FDA has approved a new immunotherapeutic compound known as Ipilimumab (anti-CTLA-4 antibody) [8,11], which has shown promising results in clinical trials. However, this antibody can produce toxicity and is not operative in direct melanoma antigen recognition. Although there are ongoing clinical trials aimed at examining the effectiveness of various other immunotherapeutic compounds, research on melanoma vaccines, used along with agents that optimize the immune response, is still needed and remains a viable option to treat melanoma as numerous melanoma associated antigens (MAAs) have been identified, characterized both in vivo and in vitro, and cloned since the 1970's [7,8,12,13].
The majority of melanoma associated antigens characterized thus far are proteins associated with normal physiologic processes intrinsic to melanocytes, such as melanin synthesis and melanosomal maturation [14]. Examples of proteins associated with melanin synthesis are tyrosinase, TRP-1 (gp75), and TRP-2 while proteins associated with melanosomal maturation are MART-1 and gp100 (Pmel17). In addition to these antigens, proteins over expressed and present on the cell surface of melanoma cells, such as CD146, CD71, and melanotransferrin (p97), are associated with melanoma cell aggressiveness and metastasis [15-17] Lastly, one group of antigens that remains a target of immunotherapy because of their high immunogenicity are the members of the cancer testis antigens, such as MAGE-A1 and NY-ESO-1. Although the specific functions of these antigens with respect to melanoma transformation remains to be elucidated, MAGE-A1 is known to bind to the protein Ski interacting protein (SKIP) and HDAC1. This protein complex in turn serves to repress transcription, which may be associated with the transformation process in melanoma [18,19].
Despite this elaborate characterization, not all MAAs elicit an optimal immune response, some of which may be attributed to the fact that the antigens chosen for clinical trials were not HLA typed to match the vaccinated individuals or that an adjuvant was needed to help prime an antigen specific immune response [20]. Examples of antigens used in clinical trials are gp100, tryosinase, MAGE-3, and MART-1 [21-23] but phase I and II studies have not shown promising results when administered as a single antigen/protein containing vaccine [24,25]. In addition, clinical trials have focused on using multivalent vaccines administered with immuno activating cytokines, such as IL-2 or GM-CSF [26-27]. One such clinical trial observed a high response rate and progression free survival in advanced melanoma individuals when vaccinated with gp100 and IL-2 [28]. Undoubtedly, the approach of using a multivalent MAA vaccine is much more promising, with respect to generating a melanoma specific immune response, but additional research is needed in defining the conditions that will foster a productive immune response to a specific patients melanoma cells. To that end, our group has been involved in generating a multivalent immunotherapeutic melanoma vaccine consisting of various MAAs. The uniqueness of our immunotherapeutic vaccine arises from infecting primary melanoma cells with live vaccinia virus, which acts as a natural adjuvant. However, the antigens incorporated within this immunotherapeutic vaccine need further characterization.
The principal of our multivalent immunotherapeutic vaccine relies on the use of five established patient derived primary melanoma cells (MEL-2, MEL-V, 3MM, KFM, and GLM-2). These cells have been characterized with respect to relevant melanoma associated antigen expression such as gp100, MART-1, NY-ESO-1, MAGE-A1, Tyrosinase, TRP-1 (gp75), TRP-2 as well as antigens essential to melanoma cell aggressiveness and metastasis, such as CD146, CD71, and Melanotransferrin. In addition, the five cells were also characterized as to their migratory/invasive potential in vitro as well as to their tumorigenic properties in vivo using BALB/c nude mice. Characterization of these cells will help not only in designing an effective immunotherapeutic vaccine but also to establish models, based on the migratory, invasive, and tumorigenic properties, that can be used to test anti-melanoma treatments.
MATERIALS AND METHODS
Cell Culture
Five primary allogeneic melanoma cells (MEL-2, MEL-V, 3MM, KFM, and GLM-2) were characterized in our study. MEL-V was isolated from a Caucasian male patient with recurrent metastases to the tibia and MEL-2 was isolated from a Caucasian female patient with axillary lymph node metastatic melanoma. KFM and 3MM were isolated from female patients and GLM-2 isolated from a male patient. KFM, 3MM and GLM-2 were all obtained from lymph node metastases. For all the primary cells, the tumor specimens were excised under aseptic conditions and single cell suspensions were obtained by mechanical disruption followed by filtering the homogenate over a nylon sieve. In addition, these five cells are novel to our laboratory and have already been accepted by the FDA for vaccine production. All five primary melanoma cells were cultured and maintained in RPMI supplemented with 10% fetal bovine serum (FBS) (Atlanta Biologicals, Lawrenceville, GA), 2mM L-glutamine (Mediatech), and Primocin™ 100μg/ml (Invivogen). In addition, all five primary melanoma cells were tested and determined to be free from mycoplasma as well as free from human and animal viruses. SK-Mel-28, SK-Mel-37, and SK-Mel-103 are established human melanoma cell line, which were generously gifted to us by Jedd D. Wolchok, MD, PhD (Memorial Sloan-Kettering Cancer Center, New york, NY) and were cultured in RPMI supplemented with 10% fetal bovine serum (FBS) (Atlanta Biologicals, Lawrenceville, GA), 2mM L-glutamine (Mediatech), penicillin 10,000IU/ml, streptomycin 10,000ug/ml, and Normocin™ 100μg/ml (Invivogen).
Western Blot
Protein extracts were prepared from MEL-2, MEL-V, 3MM, KFM, and GLM-2 using RIPA buffer [50 mM Tris-HCl, pH7.4, 150 mM NaCl, 0.2% sodium deoxycholate, 0.1% SDS, 0.5% NP-40] containing HALT protease/phosphatase inhibitor cocktail (Pierce(Pierce, Rockford, IL, USA). The samples were kept on ice for 30 min, and vortexed intermittently. Cell lysates were centrifuged at 4oC for 20 min at 14,000 rpm Protein concentration was determined by Bio-Rad Protein Assay (Bio-Rad, Hercules, CA, USA) [29,30]. Ten (10µg) of protein from each cell line was resolved by 12% SDS-PAGE under reducing conditions (presence of β-mercaptoethanol). Briefly, the proteins were transferred to Immobilon-P membranes (Millipore, Billerica, MA ) at 220 mA for 2hrs and membranes were blocked with 4% dried milk in TBST [200mM Tris-HCl, pH 7.4, 150mM NaCl, and 0.1% Tween-20 added fresh/liter of 1×TBS (TBS-T)] for at least 2-3 hrs on a shaker at room temperature. Subsequently, the membranes were incubated overnight at 4°C with either Gp100 (Pmel17) (ABCAM, Cambridge, MA), MART-1 (Santa Cruz Biotechnology, Santa Cruz, CA), MAGE-A1 (Santacruz Biotechnology), NY-ESO-1 (Santacruz Biotechnology), Tyrosinase (Santacruz Biotechnology), Trp-1(gp75) (Santacruz Biotechnology), Trp-2 (Santacruz Biotechnology), (Melanotransferrin (Santacruz Biotechnology), CD146 (ABCAM), CD71 (Santacruz Biotechnology ) or Actin (Santacruz Biotechnology) antibodies. All primary antibodies were diluted at 1:500 in TBS-T containing 4% milk on a shaker. Membranes were then washed three times with TBS-T and then incubated with the respective HRP labeled secondary antibody for 2hrs at room temperature on a shaker. After four washes with TBS-T and one wash with TBS, membranes were developed by ECL (Pierce) and detected on HyBlot CL™ autoradiography film (Denville Scientific, Inc, Metuchen, NJ).
Transwell Migration Assay
BD Biocoat Control Inserts (BD Biosciences, Bedford, MA) with 8-µm pore membrane filters were used for the migration assay as previously described [29,30]. Cells were harvested by trypsinization and 2.5 X 104 cells were seeded in the upper chamber in 500 µl of media containing 1% FBS. The lower chamber contained 750 µl of media supplemented with 5% FBS. After 18 hours of incubation, the non migrating cells were removed from the upper surface of the membrane by gently scrubbing using cotton tipped swab. Cells on the lower surface of the membrane were then fixed using methanol and stained using 1% toluidine blue 1% borax stain followed by two washes with distilled water. Inserts were then allowed to air dry and counted in 10X field. Data are expressed as numbers of migrated cells per 10X field micrograph for each sample well and normalized to cell counts obtained from the untreated control.
Invasion assay
Invasion assay was performed as previously described [29,30] using BD Biocoat growth Factor Reduced Matrigel Invasion chambers (BD Biosciences, Bedford, MA) with 8-µm pore membrane filters which were coated with matrigel. The protocol was essentially the same as the migration assay described above except that growth factor reduced matrigel invasion chambers were rehydrated for 2 hours using serum free RPMI media at 37°C prior to loading cells onto inserts. Once rehydrated, 2.5 X1 04 cells were resuspended in RPMI (500µl) containing 1% FBS and were carefully transferred onto the upper surface of filters in the chamber. Cells were allowed to invade for 18 hours after which cells were stained and counted similarly as described for migration assay. Percent invasion (invasion index) was calculated based on the percent of cells invading through the growth factor reduced matrigel invasion chambers relative to the cells migrating through control membrane.
Animal Studies
Tumorigenic studies on all five primary cells were performed under an approved IACUC protocol following New York Medical College institutional policy and guidelines. BALB/c nu/nu mice were purchased from The Jackson Laboratory (Bar Harbor, ME) and allowed to acclimate for one week before being injected subcutaneously with either 2 X 106 or 5 X 106 cells. All cell lines were re-suspended in 100µl. Prior to being injected with tumor cells, the mice were anesthetized by 3% isoflurane inhalation and their skin was disinfected with betadine to avoid infection. Upon tumor growth, the animals were euthanized and the excised tumors were snap frozen in liquid nitrogen and subsequently stored at -80oC for further analyses.
Generation of Tumor Tissue Protein Lysates
Tumor tissue from all four cells were finely ground using a mortar and pestel on liquid nitrogen. RIPA buffer, containing HALT protease/phosphatase inhibitor cocktail (Pierce), was added to the tissue samples followed by gentle homogenization using a Brinkmann Homogenizer for 30 seconds while on ice. The samples were then kept on ice for 30 min, and vortexed intermittently. Cell lysates were then centrifuged at 4oC for 20 min at 14,000 rpm. Protein concentration was determined by Bio-Rad Protein Assay (Bio-Rad, Hercules, CA, USA). Ten (10µg) of protein from each cell line was resolved by 12% SDS-PAGE under reducing conditions (presence of β-mercaptoethanol). Western blot for the various antigens described was performed as detailed above.
RESULTS
Primary melanoma cell lines are a source of various melanoma associated antigens. Multi-valent immunotherapeutic vaccines targeting melanoma require the use of various MAAs such that a productive immune response is initiated. The five primary melanoma cells characterized in this study expressed various MAAs as determined by Western blot analyses. The MAAs assayed for can be represented in three distinct groups based on function and cellular localization. The first group consists of antigens classically known to be over expressed in melanoma namely, MART-1, gp100 (Pmel17), MAGE-A1, NY-ESO-1 (Figure 1A). The second group of antigens is also known to be over expressed in melanoma but is involved in melanin synthesis. These antigens are tyrosinase, Trp-1 (gp75), and TRP-2 (Figure 1B). The third and last group of antigens is CD146, CD71, and melanotransferrin (Figure 1C), which are all cell surface expressed and critical to melanoma cell motility and progression [15-17] and hence critical to include in any melanoma vaccine. Collectively, the antigen groups described above have all been associated with melanosomal transformation [15-17, 31-35]. Based on the Western blot analyses, all of the cells displayed a distinct profile for the various antigens. Interestingly, gp100 was only expressed by GLM-2 while the cancer testis antigen, NY-ESO-1, was only expressed by 3MM (Figure 1A). As far as the melanoma associated antigen involved in melanin synthesis, all of the primary cells expressed tyrosinase, TRP-1, and TRP-2 (Figure 1B). The cell surface antigens, CD71 and CD146, were expressed by all of the primary cells except for GLM-2, which did not express CD71 (Figure 1C). Lastly, the cell surface antigen, melanotransferrin (p97), was only expressed by MEL-2 (Figure 1C). The band of interest is indicated by the arrow(s) next to each blot. For TRP-1 and TRP-2 (Fig. 1B-B and 1B-C), two arrows are shown as the antibody used detects multiple forms of TRP-1 and TRP-2 due to glycosylation, which affects the proteins molecular weight. Relative expression for all the antigens, corresponding to their respective Western blot, was obtained by densitometry and represented in the bar charts present in figure 1A-1C. The cells were scored for melanoma associated antigens and represented in table I. For each marker, + indicates low expression, ++ indicates intermediate expression, and +++ indicates high expression. This system of characterizing the antigen expression was agreed to by all the authors and was determined by the densitometry values calculated based on the expression level of each antigen with respect to its actin expression level. As a positive control, Western blot analyses were also performed on the established cell lines SK-Mel-28, SK-Mel-37, and SK-Mel-103.
Table I
Expression of Melanoma Associated Antigens in Primary and Established Cell Lines.
| Gp100 (Pmel17) | MART-1 | MAGE-A1 | NY-ESO-1 | Tyrosinase | TRP-1 (Gp75) | TRP-2 | Melanotransferrin (p97) | CD71 | CD146 | |
|---|---|---|---|---|---|---|---|---|---|---|
| MEL-2 | - | ++ | - | - | +++ | +++ | +++ | + | + | +++ |
| MEL-V | - | - | + | - | + | +++ | + | - | ++ | +++ |
| 3MM | - | - | - | + | ++ | +++ | + | - | + | - |
| KFM | - | ++ | ++ | - | ++ | ++ | ++ | - | +++ | + |
| GLM-2 | +++ | +++ | - | - | +++ | +++ | ++ | - | - | ++ |
| SK-Mel-28 | + | +++ | - | +++ | ++ | +++ | +++ | +++ | ++ | +++ |
| SK-Mel-37 | - | - | +++ | - | ++ | + | ++ | ++ | +++ | +++ |
| SK-Mel-103 | - | - | - | - | + | + | ++ | - | - | - |
Fig 1
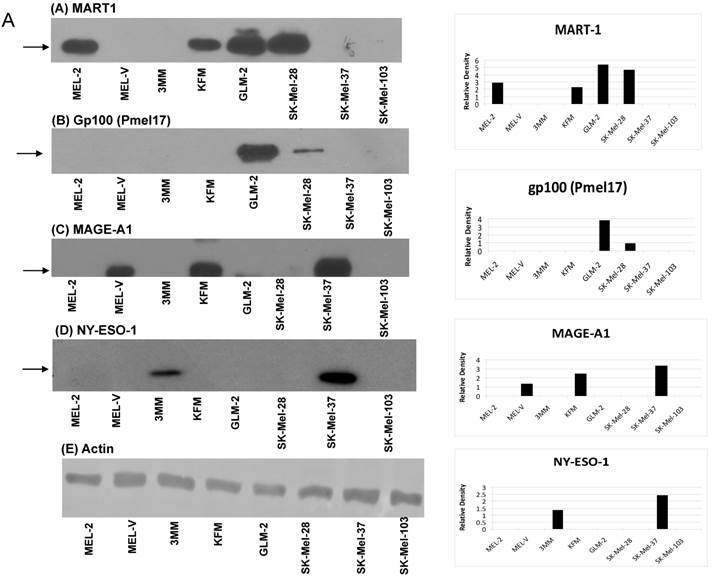
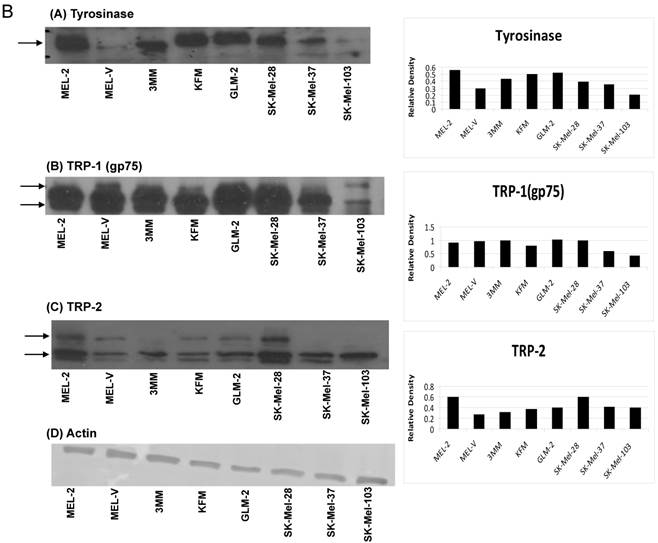
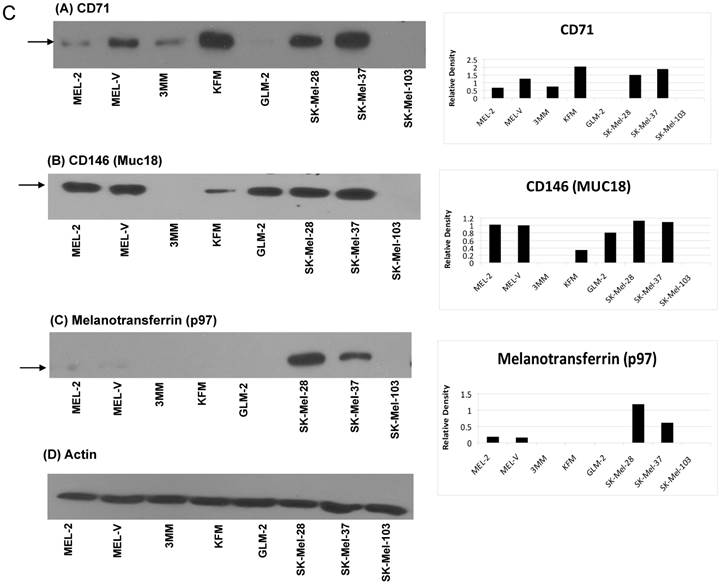
Differential expression of melanoma associated antigens. Whole cell protein lysates were prepared from all five primary melanoma cell lines (MEL-2, MEL-2, 3MM, KFM, and GLM-2) and three established melanoma cell lines (SK-Mel-28, SK-Mel-37, and SK-Mel-103). Ten micrograms (10 μg) of protein were resolved by SDS-PAGE and subsequently analyzed by Western blot analysis for antigen expression. All melanoma cell lines have differential expression of various antigens classically known to be over expressed in melanoma such as gp100 (Pmel17), MART-1, MAGE-A1 and NY-ESO-1 (Fig.1A), as well as antigens involved in melanin synthesis, such as Tyrosinase, Trp-1 (gp75), and TRP-2 (Fig. 1B). In addition, cell surface antigens not classically know as melanoma associated (MAAs) but over expressed in melanoma were assayed for such as CD71, CD146, and melanotransferrin (Fig. 1C). The band of interest is indicated by the arrow(s) next to each blot. For TRP-1 and TRP-2 (Fig. 1B-B and 1B-C), two arrows are shown as the antibody used detects multiple forms of TRP-1 and TRP-2 due to glycosylation, which affects the proteins molecular weight. The relative expression levels for each antigen was calculated by densitometry based on the actin expression levels for each antigen and is represented by the bar charts shown in figures 1A-1C.
Primary melanomas cell lines have varying migratory and invasive properties in vitro. Migration and invasion are two properties of cancer cells that are indispensable for tumor growth and metastasis [36]. All five primary cells were obtained from metastatic foci of patients diagnosed with melanoma, which supports the fact that these cells have intrinsic properties enabling them to migrate from the primary tumor mass and invade secondary sites thus establishing metastatic foci. Our results indicate that MEL-2, MEL-V, 3MM, KFM, and GLM-2 were capable of migrating, some more than others. Specifically, MEL-2 had the greatest migratory rate compared to MEL-V, 3MM, KFM, and GLM-2 (Figure 2). When comparing each cell line for its invasive properties, represented as the invasion index, GLM-2 had the highest invasion index followed MEL-2. MEL-V, 3MM, and KFM had comparable invasion indices (Figure 3).
Primary melanoma cells grow in vivo. The ability of cell lines to grow and form tumors in vivo provides a valuable tool by which treatments targeting tumors, such as chemical compounds, can be assayed for. All five primary cells characterized in this study were assayed for their tumorigenic property in vivo by injecting BALB/c nu/nu mice with the MEL-2, MEL-V, 3MM, KFM, and GLM-2 cell lines. Three BALB/c nu/nu mice were used per cell. Of the five primary cells, only MEL-2, KFM, and GLM-2 were capable of growing in vivo and forming solid subcutaneous tumors when injected with a concentration of 2 X 106 cells (Figure 4A-C). MEL-V and 3MM failed to grow in vivo using the same number of cells but when the cell number was increased to 5 X 106, only MEL-V was capable of growing in vivo (Figure 4D). The tumor measurements in diameter for all four cells ranged from 70 mm2 to 180 mm3 and are shown in table II. In addition, there was a latency period of about 2 weeks from the time tumor cells were subcutaneously injected to observing a palpable tumor mass for MEL-2, KFM, and GLM-2. MEL-V had a latency period of about one month from the time cells were injected to observing a palpable tumor. It is noteworthy to mention that of all the five melanoma cells, MEL-V has the slowest growth rate in vitro and this was also observed in vivo. Interestingly, when grown in vitro, MEL-2, KFM, and GLM-2 did not demonstrate any signs of pigmentation but produced pigmented tumors in the mice (Figure 4A-C), indicative of melanin production, as opposed to MEL-V, which did not produce a pigmented tumor (Figure 4D). This observation is most likely mediated by various factors present within the extra-cellular environment and merits further investigation.
Table II
Average Tumor Diameter.
| Cell | Tumor Measurement (mm2) |
|---|---|
| MEL-2 | 140 ± 0.26 |
| KFM | 70 ± 0.11 |
| GLM-2 | 110 ± 0.23 |
| MEL-V | 180 ± 0.25 |
Melanoma cell tumors express various melanoma associated antigens. Further extending our characterization studies on melanoma associated antigens, we wanted to determine if the primary melanoma cell xenografts have similar antigen expression when grown in vivo compared to their growth in vitro. In figures 5A-5C, each antigen was compared between the tumor tissue and the in vitro cultured cell line. The letter “C” next to the cell name denotes the in vitro cultured cells while the letter 'T” denotes the tumor tissue. The antigens assayed for in the tumor tissues were divided into three groups. The first group consists of antigens classically known to be over expressed in melanoma namely, MART-1, gp100 (Pmel17), and MAGE-A1 (Figure 5A). The second group of antigens is also known to be over expressed in melanoma but is involved in melanin synthesis. These antigens are tyrosinase, Trp-1 (gp75), and TRP-2 (Figure 5B). The third and last group of antigens that we analyzed for are CD146 and CD71 (Figure 5C). Overall, there were no significant differences in antigen expression between the cell lines and the cell line tumors (Figure 5A-5C). The band of interest is indicated by the arrow(s) next to each blot. For TRP-1 and TRP-2 (Fig. 5B-B and 5B-C), two arrows are shown as the antibody used detects multiple forms of TRP-1 and TRP-2 due to glycosylation, which affects the proteins molecular weight. Relative expression for all the antigens, corresponding to their respective Western blot, was obtained by densitometry and represented in the bar charts present in figures 5A-5C. As noted in the preceding section, MEL-V did not form pigmented tumors, which could be attributed to the fact that the MEL-V tumor had very little expression of tyrosinase, compared to the tyrosinase expression in the other tumors (MEL-2, KFM, and GLM-2).
Fig 2
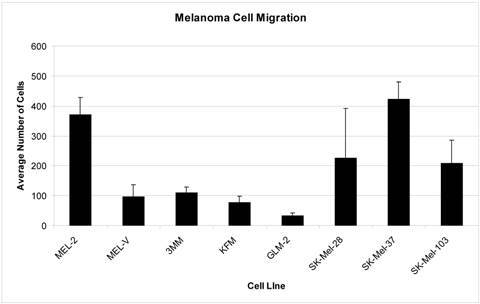
Melanoma cells migrate in vitro. Migration studies were performed using 2.5 X 104 cells per insert. The cells were seeded in the upper chamber in 500 µl of media containing 1% FBS. The lower chamber contained 750 µl of media supplemented with 5% FBS. Cells were allowed to migrate for 18 hours. All of the primary melanoma cell lines were capable of migrating through the matrigel coated inserts at varying rates with MEL-2 displaying the highest migratory rate represented as average number of cells. MEL-V, 3MM, KFM, and GLM-2 migrated at similar rates. The established melanoma cell lines (SK-Mel-28, SK-Mel-37, and SK-Mel-103) also displayed differential migratory properties as well. The data is expressed as numbers of migrated cells per 10X field micrograph for each sample well. Each experiment was repeated three times with similar results observed and the standard deviation shown above each sample represents three replicates in one experiment.
Fig 3
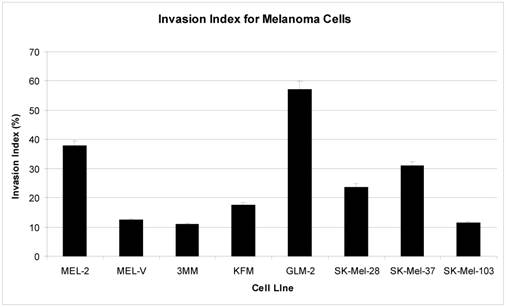
Melanoma cells display invasive properties in vitro. Melanoma cells, 2.5 X 104 cells, were resuspended in RPMI (500µl) containing 1% FBS and allowed to invade for 18 hours. All of the primary melanoma cells (MEL-2, MEL-V, 3MM, KFM, and GLM-2) displayed differential invasive properties as did the established melanoma cell (SK-Mel-28, SK-Mel-37, and SK-Mel-103). The data is represented as invasion index, which is calculated as the percent of cells invading through the growth factor reduced matrigel invasion chambers relative to the cells migrating through control membrane. Each experiment was repeated three times with similar results observed and the standard deviation shown above each sample represents three replicates in one experiment.
Fig 4
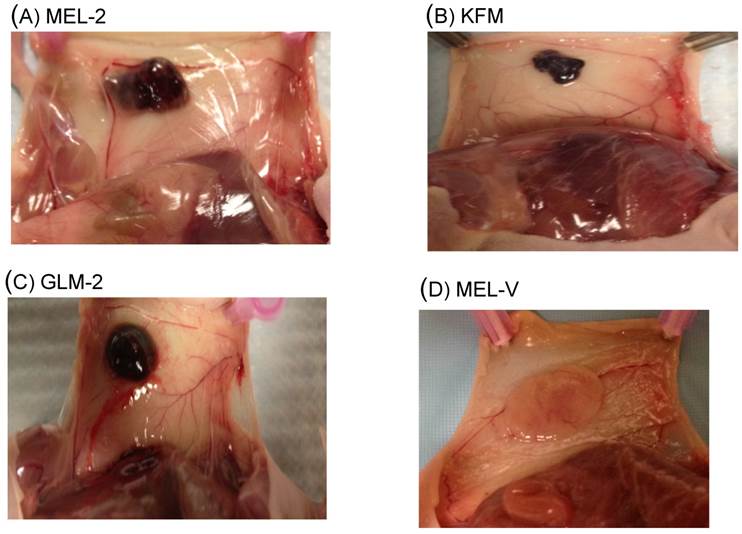
Primary melanoma cells are tumorigenic in nude mice. BALB/c nu/nu mice were anesthetized by 3% isoflurane inhalation after which 2 X 106 cells/ 100μl were injected subcutaneously. Three mice were used per cell line and tumor growth was observed in all injected mice. MEL-2, KFM, and GLM-2 were capable of forming solid pigmented tumors when injected with 2 X 106 cells (Fig. 4A-C) as opposed to MEL-V which formed solid non-pigmented tumors when injected with 5 X 106 cells (Fig. 4D).
Fig 5
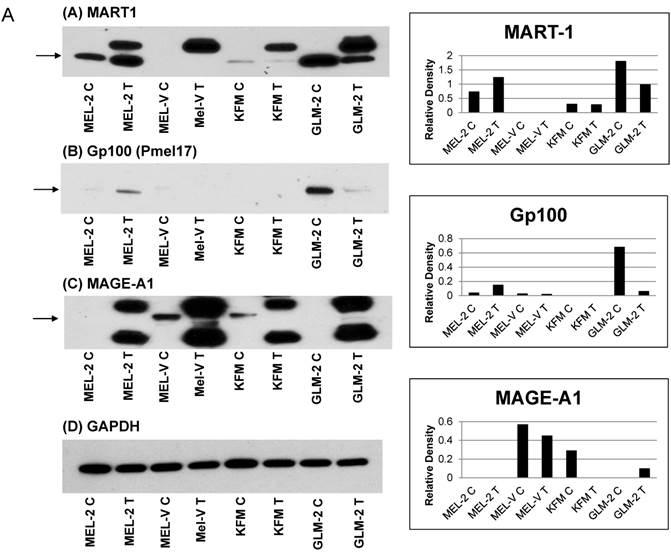
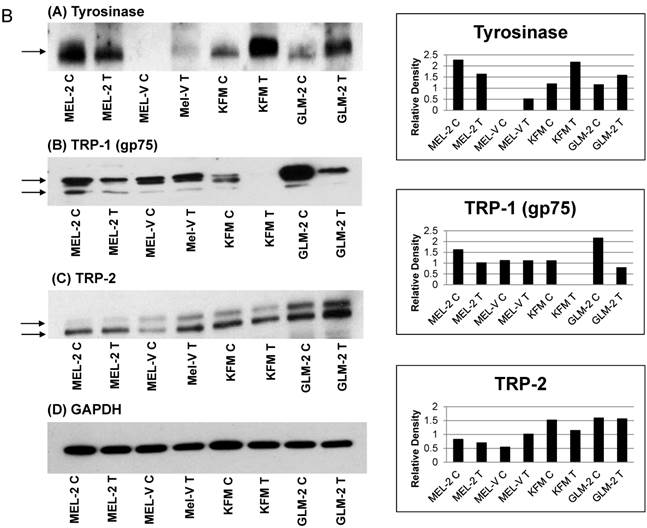
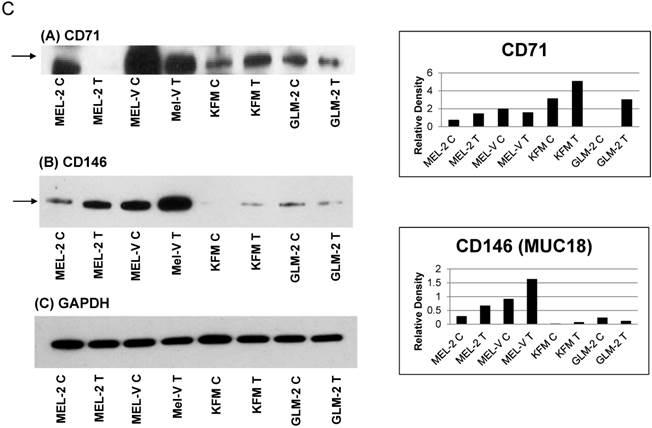
Melanoma cell tumors express various melanoma associated antigens. Melanoma cell tumor tissues were homogenized in RIPA buffer using a Brinkmann homogenizer. Ten (10µg) of protein from each tumor was resolved by 12% SDS-PAGE and subjected to Western blot analyses for the various antigens described. The letter “C” next to the cell line name denotes the in vitro cultured cell line while the letter 'T” denotes the tumor tissue. The antigens were divided into three groups. Group 1 consists antigens known to be over expressed in melanoma namely, gp100 (Pmel17), MART-1, and MAGE-A1 (Figure 5A). The second group of antigens over expressed in melanoma but are involved in melanin synthesis, such as tyrosinase, Trp-1 (gp75), and TRP-2 (Figure 5B). The third group consists of antigens, which are cell surface localized and associated with melanoma cell aggressiveness and metastasis, such as CD146 and CD71 (Figure 5C). Overall, there were no significant differences in antigen expression between the cell lines and the cell line tumors. The band of interest is indicated by the arrow(s) next to each blot. For TRP-1 and TRP-2 (Fig. 5B-B and 5B-C), two arrows are shown as the antibody used detects multiple forms of TRP-1 and TRP-2 due to glycosylation, which affects the proteins molecular weight. The relative expression for each antigen was calculated by densitometry based on the GAPDH expression for each antigen.
DISCUSSION
Melanoma, as clinically diagnosed, is pigmented in 99% of the cases suggesting that there may be a direct link of melanin synthesis and the transformation process [14]. Melanoma has been traditionally recognized as the most immunogenic of all cancers and target antigen analysis reveals that several antigens involved in melanin synthesis have been clinically exploited, which include gp100 (Pmel17) and MART-1[37-39]. In addition, there are various active clinical trials in melanoma that are focusing on; autologous melanoma cells as a vaccine, peptide based melanoma vaccine in conjunction with interferon gamma, and vaccination with gp75 in conjunction with IL-15 to name a few. However, in recent years, the clinical development of immunotherapeutic treatments for melanoma has been directed towards either augmenting an immune response to specific antigens, fine tuning an immune response by blocking inhibitory signaling molecules, such as CTLA-4 and PD1 [40,41] or evaluating combinations of both stimulatory and inhibitory responses. While the final verdict is awaited, the debate over the use of single antigens or a cocktail of various melanoma associates antigens is still ongoing. We believe that the use of multiple melanoma associated antigens combined with the removal of immune system restraints need to be tested. To this end, we have attempted to define and characterize the contribution of melanin synthesis, melanosomal maturation, and cell surface antigens to the melanoma cell phenotype using five primary melanoma cell lines.
The antigens characterized in our study can be essentially divided into three groups. The first group is composed of four antigens, MAGE-A1, NY-ESO-1, MART-1, and gp100, which have been observed to elicit immune responses in various studies [42]. These antigens from group one are either cancer testis antigens unregulated in melanoma cells (MAGE-A1 and NY-ESO-1) or antigens known to be upregulated in melanoma and function in melanosomal maturation (gp100 and MART-1). The second group consists of antigens associated with melanin synthesis, such as tyrosinase, TRP-1 and TRP-2. Lastly, group three is composed of three antigens, CD71, CD146 and Melanotransferrin, which are cell surface antigens and are markers of melanoma cell aggressiveness and metastasis [15-17]. The primary cells characterized in this study expressed the proteins required for melanin synthesis, namely tyrosinase, TRP-1, and TRP-2 however, there were variations in the expression of MART-1 and gp100 between all the cell lines with GLM-2 being the only cell line expressing both antigens at equivalent levels. Interestingly, four of the five cell lines, MEL-2, MEL-V, KFM, and GLM-2, were capable of growing in BALB/c nude mice. MEL-2, KFM, and GLM-2 cells grew in vivo when injected at a concentration of 2 X 106 cells and were also capable of producing pigmented tumors. Mel-V on the other hand, grew in vivo only when the number of cells injected was increased to 5 X 106 cells but did not produce pigmented tumors, which may be reflected in the fact that MEL-V has the lowest expression levels of tyrosinase as compared to MEL-2, KFM, and GLM-2.
In vitro, all of the primary cells did not produce melanin. Culturing of the primary cells in vitro with specific growth factors and hormones, such as basic fibroblast growth factor (bFGF) and alpha melanocyte stimulating hormone (α-MSH), failed to induce melanin synthesis. Further, conditioned media from activated stroma or co-culture of activated stromal cells with the primary cells also did not result in melanin synthesis [unpublished observations]. The use of cytokines, extracellular matrix components, and activated fibroblasts, singly or in combination, were unable to induce non-melanin producing primary cells in vitro. The idea that in vivo these cells produce melanin and that this process may be linked to tumorigenesis is thought provoking and an indicator that vaccines should incorporate antigens involved in melanin production. These observations are a reflection of the complex biology of melanin synthesis involving several cell types, which require a degree of sophistication and dynamic interchange of secretory molecules to emulate in vivo melanin synthesis in vitro [14]. These in vivo experiments however point to an underlying threshold requirement of several antigens involved in melanin synthesis and at least one antigen of melanosomal maturation for effective tumor implantation. This is significant since the majority (>99%) of human melanomas are melanin positive and as such an effective immune stimulatory therapeutic strategy would target a combination of antigens belonging to both melanin synthesis and melanosomal maturation. The underlying idea would be to decrease the threshold levels of melanin synthesis and prevent melanosome maturation.
These primary melanoma cells were obtained from metastatic foci of patients and it was incumbent on our characterization study to determine if they retained their aggressive phenotype in vitro. We utilized the Boyden chamber assay to examine the migratory and invasive properties of the five primary cells. The expression of these antigens correlated well with the invasion index as low or no expression of either CD146 or CD71 in cells such as 3MM and SK-Mel-103, had the lowest invasion index.
As evident by the expression of the various antigens in the primary cells, it is necessary to generate a multivalent antigenic response. The modality of the response, while still being investigated, in all probability should involve CD8+ T-cells in a vaccinating scenario. We are in the process of investigating the mechanism of the immune response using defined antigens singly and in combination. One should also be aware that the real evidence would be in the clinical trial as animal models have their limitations. The original clinical experimentation conducted by our group was done using vaccinia virus, which presumably acts as a potent adjuvant. Under the present scenario, the use of GM-CSF as an adjuvant for an optimal response would be a great choice as it is known to have very limited systemic reactions.
Our studies clearly highlighted the heterogeneous nature of melanoma associated antigens in primary cells derived from metastatic lesions. The plasticity and adaptability of tumor cells to switch from melanin producers in vivo to non-producers in vitro expose the need for 3-dimensional tissue re-structuring and use of these cells for screening of small molecule inhibitors and development of multivalent immunotherapeutic vaccines. It is essential that melanoma specific antigens for an immunotherapeutic vaccine be selected based on various biological properties of melanoma cells such as melanin synthesis, melanosomal maturation as well as cell surface antigens which confer an invasive phenotype on cancer cells. We would hypothesize that combinations of several melanin synthesis antigens, together with one melanosomal maturation antigen and an antigen associated with metastasis should be part of the immune stimulatory combination. The antigens described here are only representative although not exhaustive but their grouping in category is significant. In a human trial the presence of these antigens can be determined in the primary tumor for a secondary prevention trial or an immunotherapeutic trial with low tumor burden. In an ideal treatment strategy, for a sustained immune response and optimal targeted immune stimulation, the immune stimulatory antigen combination should be supplemented by the addition of anti-CTLA4 or PD-1 such that host tolerance to the antigens can be overcome. Specific animal models to examine this approach are being presently developed in the laboratory, which will help us to define the multivalent immune response using an experimental humanized model system.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Bandarchi B, Linglei M, Navab R. et al. From Melanocyte to Metastatic Malignant Melanoma. Dermatol Res Pract. 2010 583748
2. Atkins MB, Hsu J, Lee S. et al. Phase III trial comparing concurrent biochemotherapy with cisplatin, vinblastine, dacarbazine, interleukin-2, and interferon alfa-2b with cisplatin, vinblastine, and dacarbazine alone in patients with metastatic malignant melanoma (E3695): a trial coordinated by the Eastern Cooperative Oncology Group. J Clin Oncol. 2008;26:5748-5754
3. Bedikian AY, Millward M, Pehamberger H. et al. Bcl-2 antisense (oblimersen sodium) plus dacarbazine in patients with advanced melanoma: the Oblimersen Melanoma Study Group. J Clin Oncol. 2006;24:4738-4745
4. Weber J. Immunotherapy for melanoma. Curr Opin Oncol. 2011;23:163-169
5. Block MS, Suman VJ, Nevala WK. et al. Pilot study of granulocyte-macrophage colony-stimulating factor and interleukin-2 as immune adjuvants for a melanoma peptide vaccine. Melanoma Res. 2011;21:438-445
6. McCardle TW, Messina JL, Sondak VK. Completely regressed cutaneous melanocytic lesion revisited. Semin Oncol. 2009;36:498-503
7. Overwijk WW, Restifo NP. Autoimmunity and the immunotherapy of cancer: targeting the "self" to destroy the "other". Crit Rev Immunol. 2000;20:433-450
8. Pittet MJ, Zippelius A, Valmori D. et al. Melan-A/MART-1-specific CD8 T cells: from thymus to tumor. Trends Immunol. 2002;23:325-328
9. Gnjatic S, Nishikawa H, Jungbluth AA. et al. NY-ESO-1: review of an immunogenic tumor antigen. Adv Cancer Res. 2006;95:1-30
10. Hodi FS, O'Day SJ, McDermott DF. et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711-723
11. Flaherty KT, Yasothan U, Kirkpatrick P. Vemurafenib. Nat Rev Drug Discov. 2011;10:811-812
12. Boon T. Tumor antigens recognized by cytolytic T lymphocytes: present perspectives for specific immunotherapy. Int J Cancer. 1993;54:177-180
13. Rosenberg SA. A new era of cancer immunotherapy: converting theory to performance. CA Cancer J Clin. 1999;49:70-73
14. Wasmeier C, Hume AN, Bolasco G. et al. Melanosomes at a glance. J Cell Sci. 2008;121:3995-3999
15. Luo Y, Zheng C, Zhang J. et al. Recognition of CD146 as an ERM-binding protein offers novel mechanisms for melanoma cell migration. Oncogene. 2012;31:306-321
16. van Muijen GN, Ruiter DJ, Hoefakker S. et al. Monoclonal antibody PAL-M1 recognizes the transferrin receptor and is a progression marker in melanocytic lesions. J Invest Dermatol. 1990;95:65-9
17. Roland Y, Demeule M, Fenart L. et al. Inhibition of melanoma brain metastasis by targeting melanotransferrin at the cell surface. Pigment Cell Melanoma Res. 2009;2:86-98
18. Gjerstorff MF, Kock K, Nielsen O. et al. MAGE-A1, GAGE and NY-ESO-1 cancer/testis antigen expression during human gonadal development. Hum Reprod. 2007;22:953-60
19. Laduron S, Deplus R, Zhou S. et al. MAGE-A1 interacts with adaptor SKIP and the deacetylase HDAC1 to repress transcription. Nucleic Acids Res. 2004;32:4340-4350
20. Ohnmacht GA, Marincola FM. Heterogeneity in expression of human leukocyte antigens and melanoma-associated antigens in advanced melanoma. J Cell Physiol. 2000;182:332-338
21. Reynolds SR, Celis E, Sette A. et al. Identification of HLA-A*03, A*11 and B*07-restricted melanoma-associated peptides that are immunogenic in vivo by vaccine-induced immune response (VIIR) analysis. J Immunol Methods. 2000;244:59-67
22. Reynolds SR, Celis E, Sette A. et al. HLA-independent heterogeneity of CD8+ T cell responses to MAGE-3, Melan-A/MART-1, gp100, tyrosinase, MC1R, and TRP-2 in vaccine-treated melanoma patients. J Immunol. 1998;161:6970-6976
23. Reynolds SR, Oratz R, Shapiro RL. et al. Stimulation of CD8+ T cell responses to MAGE-3 and Melan A/MART-1 by immunization to a polyvalent melanoma vaccine. Int J Cancer. 1997;72:972-976
24. Baurain J, Stas M, Hammouch F. et al. Association of primary melanoma ulceration and clinical benefit of adjuvant vaccination with tumor-specific antigenic peptides. J Clin Oncol. 2009;27:15
25. Kruit WH, van Ojik HH, Brichard VG. et al. Phase 1/2 study of subcutaneous and intradermal immunization with a recombinant MAGE-3 protein in patients with detectable metastatic melanoma. Int J Cancer. 2005;117:596-604
26. Slingluff CL Jr, Petroni GR, Olson WC. et al. Effect of granulocyte/macrophage colony-stimulating factor on circulating CD8+ and CD4+ T-cell responses to a multipeptide melanoma vaccine: outcome of a multicenter randomized trial. Clin Cancer Res. 2009;15:7036-7044
27. Filipazzi P, Pilla L, Patuzzo R. et al. Adjuvant multipeptide vaccination in high-risk early melanoma patients. J Clin Oncol. 2008;26:3014
28. Schwartzentruber DJ, Lawson DH, Richards JM. et al. gp100 peptide vaccine and interleukin-2 in patients with advanced melanoma. N Engl J Med. 2011;364:2119-2127
29. Rajoria S, Suriano R, George A. et al. Estrogen induced metastatic modulators MMP-2 and MMP-9 are targets of 3,3'-diindolylmethane in thyroid cancer. PLoS One. 2011;6:e15879
30. Rajoria S, Suriano R, Shanmugam A. et al. Metastatic phenotype is regulated by estrogen in thyroid cells. Thyroid. 2010;20:33-41
31. Barrow C, Browning J, MacGregor D. et al. Tumor antigen expression in melanoma varies according to antigen and stage. Clin Cancer Res. 2006;12:764-771
32. Hoashi T, Watabe H, Muller J. et al. MART-1 is required for the function of the melanosomal matrix protein PMEL17/GP100 and the maturation of melanosomes. J Biol Chem. 2005;280:14006-40016
33. Wang RF, Appella E, Kawakami Y. et al. Identification of TRP-2 as a human tumor antigen recognized by cytotoxic T lymphocytes. J Exp Med. 1996;184:2207-2216
34. Robbins PF, el-Gamil M, Kawakami Y. et al. Recognition of tyrosinase by tumor-infiltrating lymphocytes from a patient responding to immunotherapy. Cancer Res. 1994;54:3124-126
35. Wang RF, Robbins PF, Kawakami Y. et al. Identification of a gene encoding a melanoma tumor antigen recognized by HLA-A31-restricted tumor-infiltrating lymphocytes. J Exp Med. 1995;181:799-804
36. Rajoria S, Suriano R, George AL. et al. Estrogen activity as a preventive and therapeutic target in thyroid cancer. Biomed Pharmacother. 2012;66:151-8
37. Tarhini AA, Leng S, Moschos SJ. et al. Safety and immunogenicity of vaccination with MART-1 (26-35, 27L), gp100 (209-217, 210M), and tyrosinase (368-376, 370D) in adjuvant with PF-3512676 and GM-CSF in metastatic melanoma. J Immunother. 2012;35:359-366
38. Sabbatini P, Tsuji T, Ferran L. et al. Phase I trial of overlapping long peptides from a tumor self-antigen and poly-ICLC shows rapid induction of integrated immune response in ovarian cancer patients. Clin Cancer Res. 2012;18:6497-508
39. Schaefer C, Butterfield LH, Lee S. et al. Function but not phenotype of melanoma peptide-specific CD8(+) T cells correlate with survival in a multiepitope peptide vaccine trial (ECOG 1696). Int J Cancer. 2012;131:874-84
40. Simeone E, Ascierto PA. Immunomodulating antibodies in the treatment of metastatic melanoma: the experience with anti-CTLA-4, anti-CD137, and anti-PD1. J Immunotoxicol. 2012;9:241-7
41. Weber J. Immunotherapy for melanoma. Curr Opin Oncol. 2011;23:163-9
42. Rosenberg SA, Zhai Y, Yang JC. et al. Immunizing patients with metastatic melanoma using recombinant adenoviruses encoding MART-1 or gp100 melanoma antigens. J Natl Cancer Inst. 1998;90:1894-900
Author contact
![]() Corresponding author: Dr. Raj K.Tiwari, Tel: (914) 594-4870 Fax: (914) 594-4879 E-mail: raj_tiwariedu.
Corresponding author: Dr. Raj K.Tiwari, Tel: (914) 594-4870 Fax: (914) 594-4879 E-mail: raj_tiwariedu.
Citation styles
APA
Suriano, R., Rajoria, S., L.George, A., Geliebter, J., Wallack, M., Tiwari, R.K. (2013). Ex Vivo Derived Primary Melanoma Cells: Implications for Immunotherapeutic Vaccines. Journal of Cancer, 4(5), 371-382. https://doi.org/10.7150/jca.6625.
ACS
Suriano, R.; Rajoria, S.; L.George, A.; Geliebter, J.; Wallack, M.; Tiwari, R.K. Ex Vivo Derived Primary Melanoma Cells: Implications for Immunotherapeutic Vaccines. J. Cancer 2013, 4 (5), 371-382. DOI: 10.7150/jca.6625.
NLM
Suriano R, Rajoria S, L.George A, Geliebter J, Wallack M, Tiwari RK. Ex Vivo Derived Primary Melanoma Cells: Implications for Immunotherapeutic Vaccines. J Cancer 2013; 4(5):371-382. doi:10.7150/jca.6625. https://www.jcancer.org/v04p0371.htm
CSE
Suriano R, Rajoria S, L.George A, Geliebter J, Wallack M, Tiwari RK. 2013. Ex Vivo Derived Primary Melanoma Cells: Implications for Immunotherapeutic Vaccines. J Cancer. 4(5):371-382.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) License. See http://ivyspring.com/terms for full terms and conditions.