Journal of Cancer
ISSN: 1837-9664
3.2
Impact Factor
ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2013; 4(2):104-116. doi:10.7150/jca.5002 This issue Cite
Research Paper
In Vitro Assessment of the Inflammatory Breast Cancer Cell Line SUM 149: Discovery of 2 Single Nucleotide Polymorphisms in the RNase L Gene
Brandon T. Nokes1, Heather E. Cunliffe2, Bonnie LaFleur3, David W. Mount1,4, Robert B. Livingston1,5, Bernard W. Futscher1,6, Julie E. Lang7 ![]()
1. University of Arizona Cancer Center;
2. Translational Genomics Research Institute, Phoenix, Arizona;
3. Division of Epidemiology and Biostatistics;
4. Bioinformatics Shared Services;
5. Department of Medicine, Division of Hematology and Oncology, University of Arizona College of Medicine, Tucson, Arizona;
6. Department of Pharmacology and Toxicology, University of Arizona College of Medicine, Tucson, Arizona;
7. Norris Comprehensive Cancer Center, University of Southern California, Los Angeles, California.
Received 2012-8-8; Accepted 2012-11-12; Published 2013-1-10
Citation:
Nokes BT, Cunliffe HE, LaFleur B, Mount DW, Livingston RB, Futscher BW, Lang JE. In Vitro Assessment of the Inflammatory Breast Cancer Cell Line SUM 149: Discovery of 2 Single Nucleotide Polymorphisms in the RNase L Gene. J Cancer 2013; 4(2):104-116. doi:10.7150/jca.5002. https://www.jcancer.org/v04p0104.htm
Other stylesAbstract
Background: Inflammatory breast cancer (IBC) is a rare, highly aggressive form of breast cancer. The mechanism of IBC carcinogenesis remains unknown. We sought to evaluate potential genetic risk factors for IBC and whether or not the IBC cell lines SUM149 and SUM190 demonstrated evidence of viral infection.
Methods: We performed single nucleotide polymorphism (SNP) genotyping for 2 variants of the ribonuclease (RNase) L gene that have been correlated with the risk of prostate cancer due to a possible viral etiology. We evaluated dose-response to treatment with interferon-alpha (IFN-α); and assayed for evidence of the putative human mammary tumor virus (HMTV, which has been implicated in IBC) in SUM149 cells. A bioinformatic analysis was performed to evaluate expression of RNase L in IBC and non-IBC.
Results: 2 of 2 IBC cell lines were homozygous for RNase L common missense variants 462 and 541; whereas 2 of 10 non-IBC cell lines were homozygous positive for the 462 variant (p= 0.09) and 0 of 10 non-IBC cell lines were homozygous positive for the 541 variant (p = 0.015). Our real-time polymerase chain reaction (RT-PCR) and Southern blot analysis for sequences of HMTV revealed no evidence of the putative viral genome.
Conclusion: We discovered 2 SNPs in the RNase L gene that were homozygously present in IBC cell lines. The 462 variant was absent in non-IBC lines. Our discovery of these SNPs present in IBC cell lines suggests a possible biomarker for risk of IBC. We found no evidence of HMTV in SUM149 cells. A query of a panel of human IBC and non-IBC samples showed no difference in RNase L expression. Further studies of the RNase L 462 and 541 variants in IBC tissues are warranted to validate our in vitro findings.
Keywords: inflammatory breast cancer, SUM149, HMTV, interferon-alpha, MMTV, RNase L.
INTRODUCTION
Inflammatory breast cancer (IBC) is a rare form of breast cancer, accounting for only 5% of breast cancer cases annually in the United States. Survival outcomes are improving for patients with non-inflammatory breast cancer (non-IBC), but remain poor for patients with IBC despite aggressive multimodal treatment 1-3. IBC, the most lethal form of breast carcinoma, is characterized by distinct clinicopathologic features, including rapid disease progression and onset of swelling, enlargement of the breast, skin tenderness, induration, edema, warmth, and erythema, which is commonly combined with peau d'orange 4-8. The 5-year survival rate for patients presenting with non-metastatic IBC is only about 40%, even with modern multidisciplinary therapy 1,7,9-12.
Treatment factors associated with improved patient survival include use of multimodality therapeutic strategies, including chemotherapy, modified radical mastectomy, and postmastectomy radiation 12-14. Although excellent rates of locoregional control are achievable 6, mortality is usually related to systemic recurrence. Because of the rarity of IBC and the inherent difficulty in obtaining tumor tissue from IBC patients, who may lack a tumor mass at presentation and typically receive upfront systemic chemotherapy, few studies have been performed to characterize its molecular biology 15-17. Understanding the distinct biologic and molecular behavior of IBC is likely to provide insight into carcinogenic mechanism(s) and aid discovery of novel targets for future treatment interventions.
Human mammary tumor virus (HMTV), a human homologue to the mouse mammary tumor virus (MMTV) has been proposed by Pogo et al. to play a role in IBC 18. MMTV predictably leads to tumor formation in mice 18; however, HMTV may or may not be associated with human cancers 18-26. Recently, Pogo et al. reported that HMTV sequences were detected in 71% of IBC cases in American women and, in turn, were associated with a more malignant breast cancer phenotype than non-IBC 26. Importantly, these findings have not been independently validated, and the significance of these putative viral DNA sequences in humans remains unclear 18-26.
The biology of IBC is distinct from that of non-IBC, in that IBC progresses much more rapidly (weeks to months, rather than months to years) and has unique clinical features, such as skin erythema, warmth, lack of a discrete mass, and often the presence of dermal lymphatic invasion. Given the markedly different clinical presentation of IBC (as compared with non-IBC) along with diverse viral etiologies implicated in other types of cancer, we hypothesized that IBC may have a viral cause, possibly involving putative HMTV infection. Given the scarcity of available human IBC tissue samples available for research at any single institution, we chose to leverage the 2 commercially available immortalized IBC cell lines SUM149 (the most widely used cell line model in IBC studies 27-29) and SUM190 as appropriate models for testing our hypothesis. Cell culture experiments were performed using SUM149 due to the extremely fastidious nature of manipulating SUM190 cells in culture.
Considering HMTV is still a putative virus, we assessed our in vitro IBC model for carrying a viral infection by multiple modalities. First, we sought to define single nucleotide polymorphisms (SNPs) for the ribonuclease (RNase) L gene, the product of which combats viral infection by degrading viral RNA and inducing apoptosis of infected cells 30-33. Several non-synonymous coding SNPs have already been associated with high risk of prostate cancer 30,31 and further reported to be associated with a putative oncogenic viral infection 32. We chose to investigate the significance of two common missense variants R462Q (rs486907) and E541D (rs627928) reported to associate with the incidence rate of sporadic prostate cancer in several studies. Given the utility of these RNase L SNPs as high-risk biomarkers for susceptibility to prostate cancer, we sought to determine whether IBC cell lines contained the same high-risk genotypes, potentially serving as an indicator of genetic susceptibility to viral infection and IBC carcinogenesis. Moreover, the RNase L gene is a downstream effector of the type 1 interferon pathway, which is utilized in mitigating viral infections (Figure 1), and altered function of this gene product may render cells more susceptible to cancer development, as the normal function of type 1 IFN-induced RNase L expression is to trigger destruction of viral RNA 30-32.
An allelic discrimination assay for the downstream interferon (IFN) effector RNase L was utilized to detect 2 SNP variants associated with cancer risk, impacting amino acids 462 and 541 34. We hypothesized that variations in genotypic frequency of these RNase L SNPs may indicate an association of increased risk of a viral infection and potential etiology within IBC vs. non-IBC cell lines. We also conducted a search on the National Center for Biotechnology Information (NCBI) Gene Expression Omnibus (GEO) website to assess differences in expression of RNase L between IBC and non-IBC tumor tissues 35.
Our second approach to determining the possibility of viral infection of IBC cells, we assayed for a selective decrease in proliferation of SUM149 cells in response to interferon-alpha (IFN-α) treatment (a naturally occurring antiviral cytokine). Interferons (IFNs) are effective molecules for studying the biology of viral cause: in vertebrates, they are produced naturally by many nucleated cells in response to viral, parasitic, and tumor-derived challenges. IFNs assist the immune response by inhibiting viral replication within host cells, activating natural killer cells, and increasing antigen presentation to lymphocytes 36,37. Moreover, the IFN-mediated 2-5A pathway is a key innate response to viral infection via RNase L-induced viral RNA degradation, as well as a mediator of apoptosis 38,39.
Lastly, to investigate findings by Pogo et al 18, we sought whether determine whether HMTV elements are present in the IBC cell line SUM149 using RT-PCR and Southern blot analysis.
Figure 1
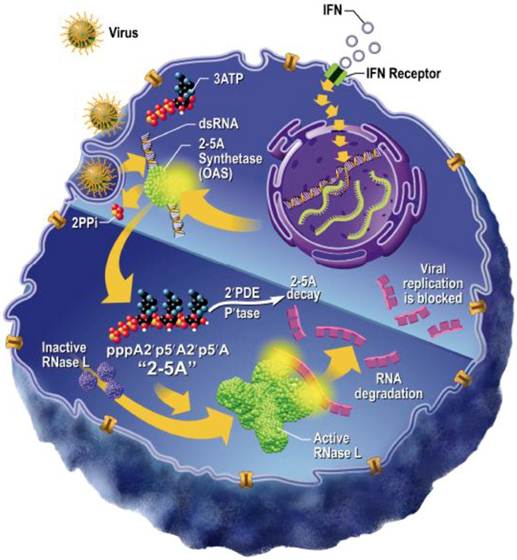
Schematic representation of the innate IFN-RNase L pathway as influenced by viral infection. Viral infection triggers the release of Type I interferons, which act on the infected cell in an autocrine fashion. IFN signaling initiated the Oligoadenylate Synthase (OAS)/RNase L pathway for nucleic acid degradation (Adapted from Silverman 2007) [50].
MATERIALS AND METHODS
Allelic Discrimination Assay We tested for the presence of RNase L variants R462Q and E541D as potential indicators of a genetic risk factor for IBC, using the TaqMan allelic discrimination assay (Life Technologies, Carlsbad, CA). Primer probes were identical to those designed by Shook et al. 31. The primers and probes for R462Q were: forward primer 5'-GGAAGATGTGGAAAATGAGGAAGA-3', reverse primer 5'-TGCA- GATCCTGGTGGGTGTA-3', and probes 5'-VIC-CAGGACATTTCGGG- CAA-MGB and 5'-FAM-CAGGACATTTTGGGCAA-MGB. The primers and probes for E541D were: forward primer 5'-TCTATGTGGTAAAGAAGGGAAGCA-3', reverse primer 5'-TTGAACCACCTCTTCATTACTTTGAG-3', and probes 5'-VIC-TTTCAGATCCT-CAAAT-MGB and 5'-FAMTTTCAGCTCCTCAAAT-MGB 31.
We extracted genomic DNA from 12 cell lines using the QIAamp DNA purification kit (Qiagen, Valencia, CA). We used 20 ng of DNA per reaction in 96-well plate format. All reactions were conducted in triplicate using an ABI 7500 Fast RT PCR System and analyzed using SDS 2.0 software (Life Technologies). P values for the cell line SNP comparisons were calculated using the online contingency table from VassarStats (http://vassarstats.net).
Bioinformatics Analysis A comparative search was performed on the NCBI GEO site (http://www.ncbi.nlm.nih.gov/geo/) in order to assess differences in expression of RNase L between IBC and non-IBC tumor tissues. The GSE5847 entry originally published by Boersma et al. provided a suitable data set with 15 IBC and 35 non-IBC tumor samples, with 2 normal breast tissue samples as a control 35. The decision to use this data set was based on lack of available entries for IBC samples, as well as the consistent nature of the expression profile of stromal tissue relative to tumor samples. Moreover, because we were looking for a DNA-based marker of genetic susceptibility, and tumor cells are highly heterogeneous, we selected the stromal data set for this analysis.
Statistical Considerations. To assess SNP prevalence between IBC and non-IBC breast stromal tissue samples, a computational script was written in the language R based on the sample size analysis recommendations made by Pfeiffer et al., 40. The script was then independently verified using the Bioinformatics Institute's (BII) Online Sample Size Estimator (http://osse.bii.a-star.edu.sg/). Using a case-control design, based on the lowest minor allele frequency (MAF) for SNP rs486907; we based these estimates on the MAF for rs486907, because the lower allele frequency will require a larger sample size. We calculated the required sample sizes to test a significant risk ratio between normal samples and IBC or non-IBC samples, as well as the risk ratio between IBC and non-IBC samples.
Cell Culture SUM149 cells, BT474 cells, and MDA-MB-231 cells were used in this study. All cell lines were acquired from the American Type Culture Collection (ATCC, Manassas, VA) except the SUM149 cell line sourced from Asterand (Detroit, MI). All cell lines were subjected to genotyping with the ABI Identifiler Assay (Life Technologies) for validation of cell line identity. SUM149 cells were grown in Ham's F-12 medium supplemented with 5% heat-inactivated fetal bovine serum (FBS) and 1% antibiotics/antimycotics (Invitrogen, Carlsbad, CA). BT474 cells were grown in Roswell Park Memorial Institute (RPMI) medium supplemented with 10% FBS and 1% antibiotics/antimycotics (Invitrogen). MDA-MB-231 cells were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% FBS and 1% antibiotics/antimycotics (Invitrogen). All cell lines were maintained in a humidified incubator with 5% CO2 atmosphere at 37o C. Cells were plated in triplicate at a density of 5,000 cells per well in 96-well tissue culture plates and grown to greater than or equal to 30% confluence at the time of treatment. Purified DNA from the SUM190 cell line was graciously provided by Dr. Cunliffe from the Translational Genomics Research Institute (TGen) in Phoenix, AZ.
IFN-α Treatment IFN-α (Imgenex, San Diego, CA) was dissolved in phosphate-buffered saline (PBS) with 5% fetal bovine serum to a final stock concentration of 100 µg/ml. Before treatment, the complete medium was removed and the cell monolayers were rinsed once with PBS. Cells were then treated with 0, 500, 1000, 2500, and 5000 U/ml of IFN-α for 24 hours and 48 hours. At each time point, we removed the IFN-α, rinsed the cells with PBS, and evaluated cell proliferation. For IFN-α block experiments, we pre-incubated the cells for 15 minutes with 1 µg/ml IFN-α specific antibody (Sigma, St. Louis, MO).
Proliferation Assays Cell proliferation was evaluated in triplicate for all treatments with the CyQUANT cell proliferation assay kit (Invitrogen), per the manufacturer's instructions. Briefly, IFN-α treatment was removed and the cells washed once with PBS. 100 µl CyQUANT cell proliferation assay working reagent was then added to the cells and incubated for 1 hour at 37o C. Emission (directly proportional to proliferation) was recorded on the BioTek FLx800 plate reader (Winooski, VT). Proliferation data was analyzed by one-way analysis of variance using the Tukey multiple comparisons test and GraphPad Prism software (GraphPad Prism Software, Inc., La Jolla, CA). All assays were performed in at least triplicate and were analyzed together at the same time point.
Primer Design and PCR Putative HMTV sequence were obtained from the National Center for Biotechnology Information website (http://www.ncbi.nlm.nih.gov/sites/entrez). Specific primers were designed to the env/LTR and late LRT regions. The primers used for PCR of the env/LTR region were as follows: 5'TCT GCG TTA CAC CAC TAC CG 3' and 5'TGA ACT CGA CCT TCC TCC TG 3'. The primers used for PCR of the late LTR region were as follows: 5'ACC TTC CTC CTG AGC CTA GC 3' and 5'TTT ATT AGC CCA ACC TTG CG 3'. For reverse-transcription polymerase chain reaction (RT-PCR), total RNA was isolated from SUM149, BT474 and MDA-MB-231 cells using the RNeasy purification kit (Qiagen) and cDNA produced using the first strand cDNA synthesis kit from MBI Fermentas (Glen Burnie, MD). To conduct PCR, we used MBI Fermentas reagents and evaluated products on a 1% Tris-borate-EDTA (TBE)/agarose gel. Gel images were acquired using a Gel Logic 200 Imaging System with Kodak 1D 3.6 software (Carestream Molecular Imaging, Rochester, NY).
Cloning and Sequencing PCR products of interest were excised from agarose gels and the DNA purified using the QIAquick gel extraction kit (Qiagen). Purified PCR products were then ligated into the pGEM-T Easy Vector (Promega, Madison, WI) overnight at 4o C and transformed into TOP10 Chemically Competent E. coli (Invitrogen). Blue-white colony selection was used to screen for recombinant plasmids containing ligated PCR fragments (recombinants resulting in disrupted β-galactosidase function, preventing metabolism of X-gal substrate). Recombinant plasmids were evaluated by Sanger sequencing, performed by the Genomic Analysis and Technology Core Facility at the BIO5 Institute at the University of Arizona (Tucson, AZ).
Western Blot Analysis Proteins were resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) on a 4% to 20% gradient minigel (Bio-Rad, Hercules, CA), using the Mini-PROTEAN 3 Cell, run at 100 V for 1.5 hours at 25° C. 15 μg of total protein from cytoplasmic/membrane extracts were resolved and transferred to nitrocellulose membranes using a Mini Trans-Blot Electrophoretic Transfer Cell (Bio-Rad). Efficient transfer of proteins was confirmed by SYPRO Ruby protein blot stain (Bio-Rad) and Kaleidoscope molecular weight markers (Bio-Rad). Nitrocellulose membranes were blocked for 2 hours in blocking buffer (4.0% bovine serum albumin [BSA], 10 mM PBS, 0.05% Triton X-100, pH 7.4) at 25° C. Membranes were next incubated with antihuman-specific rabbit monoclonal IFN receptor alpha IFNAR1 antibody (ab45172, Abcam, Cambridge, MA), diluted 1:20,000 v/v in blocking buffers overnight at 4° C with gentle agitation. Following incubation with primary antibody, membranes were washed 3 times in 10 mM PBS, 0.05% Triton X-100, pH 7.4 then incubated in donkey anti-rabbit secondary antibody with alkaline phosphatase (AP) conjugate (1:1000 v/v; Jackson ImmunoResearch, West Grove, PA) at room temperature for 1 hour. Nitrocellulose membranes were washed 3 times in 10 mM PBS, 0.05% Triton X-100 pH 7.4; and protein products visualized after 1 to 5 minutes following addition of AP substrate (MBI Fermentas). Quantification of a specific protein band was established with GelQuant.NET software provided by biochemlabsolutions.com. In the densitometry analysis, relative pixel density of the IFN receptor wells was normalized to that of β-actin.
Southern Blot Analysis 10 µg purified SUM149 genomic DNA was digested with 5U FastDigest BamHI restriction enzyme (MBI Fermentas) at 37o C for 30 minutes, then heat inactivated at 80o C for 5 minutes. DNA was subsequently extracted with an equal volume of isopropyl alcohol and resuspended in 10 µl nuclease-free water at room temperature for 15 minutes. The entire volume was electrophoresed through a 1% agarose gel in 1X TBE buffer for 6 hours at 3 V/cm (until the bromphenol blue marker reached the bottom of the gel). Halfway through the gel electrophoresis, we loaded a positive control synthetic fragment (IDT Technologies, San Diego CA) encoding for a 172 base-pair region of the HMTV env region 5' TAT GAT TTT ATC TGC GTT ACA CCA CTA CCG TAT AAT GCT TCT GAG AGC TGG GAA AGA ACC AAG GCT CAT TTA CTG GGC ATT TAA AAT AAC AAT GAG ATT TCA TAT AAC ATA CAA AAA TTA ACC AAC CTA ATT AGT GAT ATG AGC AAA CAA CAT ATT GAC GCA GTG GAC CTT A 3'. Before blotting, the gel was rinsed in deionized water, incubated in denaturing solution for 30 minutes at room temperature with shaking, rinsed again in deionized water and incubated in neutralization for 15 minutes at room temperature with shaking. We repeated this procedure and then transferred the DNA by traditional upward capillary action for 18 hours at room temperature. After transfer, the membrane was washed in 2X SSC solution to remove any residual agarose, dried at room temperature, and fixed by UV crosslinking for 2 minutes.
Probe Synthesis HMTV env DNA (500 ng) was labeled using the Biotin DecaLabel DNA Labeling Kit (MBI Fermentas). The HMTV env DNA template was combined with 5x decanucleotide reaction buffer and nuclease-free water. The tube was vortexed, pulse-spun for 5 seconds, incubated in a boiling water bath for 10 minutes, and quickly cooled on ice. Biotin Labeling Mix and 5U of Klenow fragment were added and the reaction incubated for 1 hour at 37° C. The reaction was stopped by adding 1μl 0.5M EDTA, pH 8.0. The labeled DNA was directly used for hybridization.
Hybridization and Detection The membrane was incubated in a pre-hybridization solution containing 5X SSC/5X Denhardt's, 0.5% SDS, 100 µg/ml nonspecific DNA (Sigma) at 42° C for 4 hours with agitation in a ProBlot 12 hybridization oven (Labnet International, Woodbridge, NJ). During this time, the biotin-labeled probe was denatured at 100° C for 5 minutes and chilled on ice. The denatured probe was added to the pre-hybridization solution to obtain a final probe concentration of 100 ng/ml and incubated it overnight at 42° C with shaking. After hybridization, the membrane was washed twice with 2X SSC, 0.1% SDS for 10 minutes at room temperature, then twice with 0.1X SSC, 0.1% SDS for 20 minutes at 65º C. Excess liquid was removed from the membrane by briefly placing it on filter paper. The biotin-labeled DNA was detected using the Biotin Chromogenic Detection Kit (MBI Fermentas), according to the manufacturer's directions. Color development started to be visible after 1 hour. We acquired both gel and membrane images using the Gel Logic 200 Imaging System with Kodak 1D 3.6 software (Carestream Molecular Imaging, Rochester, NY).
RESULTS
Analysis of R462 and E541 SNPs in human IBC and non-IBC cell lines
SNP genotyping was first performed for RNase L variants R462Q and E541D in 2 IBC and 10 non-IBC cell lines with results shown in Figure 2 and Table 1. The SUM149 and SUM190 IBC cell lines were homozygous G at rs486907 (homozygous Arginine at residue 462) and homozygous G at rs627928 (homozygous Glutamic acid at residue 541). The 541 GG and 462 AA genotype is the same as those previously reported to be associated with increased risk for sporadic prostate cancer 30,31. Of note, both IBC cell lines displayed 462 GG and 541 GG homozygous genotypes, of which, 462 GG is not associated with prostate cancer development, whereas 541 GG is. However, these genotypes did differ significantly from non-IBC genotypes at these residues suggesting the possibility of a novel risk allele for IBC. All but two of the non-IBC cell lines were either heterozygous or homozygous for the A allele at rs486907 (residue 462), and all 10 non-IBC cell lines were heterozygous or homozygous for the T allele at rs627928 (residue 541). Analysis by the Fisher's exact probability test yielded a two-tailed p value of 0.09 for the 462 variant and 0.015 for the 541 variant, as calculated using VassarStats (Vassarstats.net) contingency tables (Table 1).
Figure 2
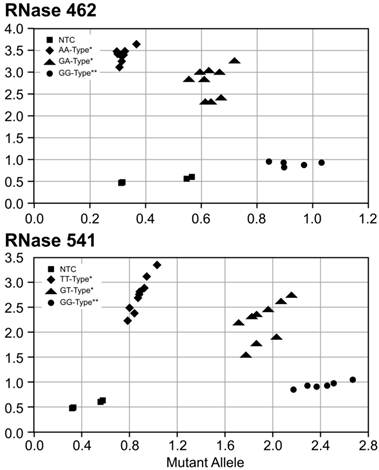
Single nucleotide polymorphism (SNP) allele discrimination scatter plot forribonuclease (RNase) L variants (A) 462 (rs486907) and (B) 541 (rs627928). The Y-axis indicates detection of the 462Q(A) and 541D (B) allele by a FAM reporter (diamonds), and the X-axis indicates detection of the 462R (A) and 541E (B) allele by a VIC reporter (circles). Heterozygous genotypes displayed amplified copies of both alleles and were found in an intermediate position between homozygous mutants and homozygous wildtype cell lines (triangles). The No Template Control (NTC) wells are denoted as squares. *Non-IBC cell lines, **Corresponds to IBC cell lines.
Table 1
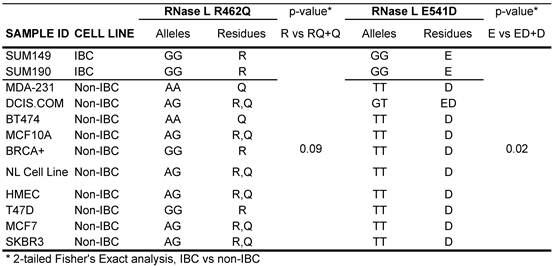
Human ancestral alleles for RNase L are “G” at SNP 462 (rs486907) and “G” at SNP 541 (rs627928). Amino acid residue abbreviations: R, Arginine; Q, Glycine; E, Glutamic Acid; D, Aspartic acid. Data from dbSNP Reference SNP build 137. The “Normal Cell Line” and “BRCA+” cell lines are both primary cell lines established by the Lang laboratory from prophylactic mastectomy specimens from individuals without breast cancer; these cell lines include both stromal and breast parenchymal components.
An evaluation of genotype frequency for these RNase L SNPs in IBC tumors is required in order to define a statistically significant correlation with potential risk of IBC in patient samples. Due to the rarity of IBC, and lack of large biorepositories for this disease, we calculated a statistical estimate of how many cases and controls would be required to perform a comprehensive analysis of this nature. The dbSNP database lists the minor allele frequency (MAF) for rs486907 to be 24% and the MAF for SNP rs627928 to be 48%. Using a case-control design, based on the MAF for SNP rs486907 (the lower allele frequency will require a larger sample size), we calculated the required sample sizes to test a significant risk ratio (estimated for a 2-fold increased risk) between healthy patient samples (without cancer) and IBC and non-IBC patient samples, as well another 2-fold risk ratio between IBC and non-IBC samples. The minimum sample size required to have 80% power to detect at least a 2-fold increase in risk between IBC and non-IBC cancer is 356 samples of each cancer; further, in order to test a difference of at least 2-fold level of risk between either IBC or non-IBC cancers to healthy individuals will necessitate 160 healthy (control) samples. This resulted in a total of 872 defined cancer type/control samples being required to evaluate the significance of these SNPs in IBC. This analysis could be accomplished should major IBC investigators choose to pool case/control resources to do so.
Our bioinformatic query using NCBI GEO data set GSE5847 regarding the differences in gene expression of RNaseL between human IBC and non-IBC samples found no significant difference in gene expression between the two groups within the stroma of human breast tissue samples (Figure 3).
Investigating the possibility of viral infection-mediated response to IFN-α treatment in SUM149 cells
We next sought to determine whether SUM149 cells showed an altered proliferative potential following IFN-α stimulation compared to non-IBC cell lines (Figure 4). We observed a dose- and time-dependent decrease in cell proliferation of SUM149 cells treated with IFN-α. At 24 hours, cell proliferation decreased by 32% relative to controls at the highest dose of 5000 U/ml IFN-α (p < 0.001). At 48 hours, cell proliferation continued to decrease at this dose, by 41% relative to controls (p < 0.01). We did not observe a corresponding decrease in SUM149 proliferation after pre-incubation of IFN-α with a specific neutralizing antibody (Figure 5, proliferation decreased by only 5% at this dose relative to controls). The results of the same INF-α response proliferation assay performed with 2 non-IBC cell lines (BT474 and MDA-MB-231) are shown in the lower panel of Figure 4. In contrast to the dose- and time-dependent decrease in cell proliferation of SUM149 cells, we did not see any decrease in cell proliferation in the MDA-MB-231 cells, and saw only a weak decrease in the BT474 cells at 24 hours that was not evident at 48 hours. Moreover, only the SUM149 cell line demonstrated a direct and specific response to IFN-α treatment (Figure 5). Note that each of the 3 cell lines expressed the IFN-α receptor IFNAR1 RNA (Figure 6A) and protein (Figure 6B), indicating that the absence of an IFN- α treatment response in the BT474 and MDA-MB-231 cell lines was not due to the lack of the receptor. The results from Western blot showed that all three cell lines expressed IR, however the expression level is much higher in SUM149 cell than the other two cell lines. Correspondingly, the relative IFNAR1 expression ratios for SUM149, MDA-MB-231 and BT474 cell lines were fold changes of 2.05 (59,601/29,037), 0.51 (107,956/212,049), and 0.17 (26,893/149,868), respectively, based on quantification of the Western blot shown in Figure 6B. Notably, there were subtle differences detected in the amount of protein loaded into each well, but this was accounted for in the densitometry analysis by normalizing each well to the amount of β-actin.
Figure 3
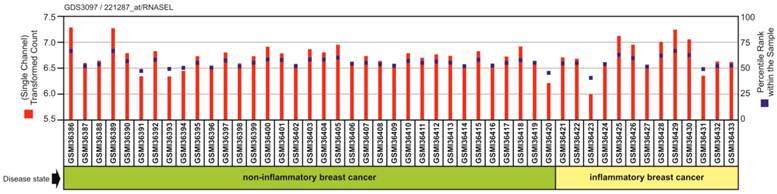
Plot of expression of gene RNASEL on Affymetrix expression microarrays of IBC and non-IBC tumor samples. This plot was generated on the NCBI GEO website from data set GSE5847 originally published by Boersma et al. 2008 [34]. Log expression value (red) and rank order of expression value (blue) are shown for each sample.
Figure 4
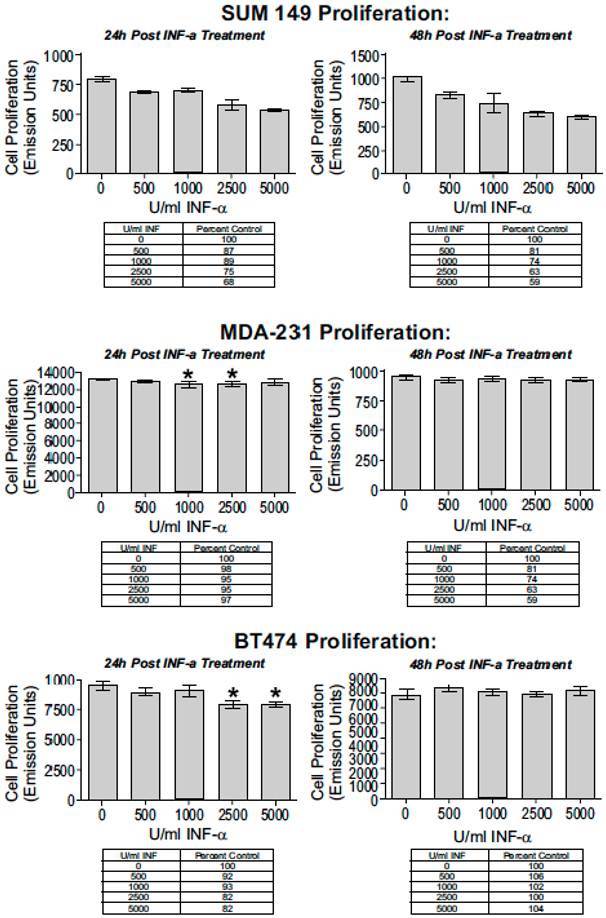
Cell proliferation as a function of Interferon (IFN- α) dosing. Increasing doses of IFN-α decreased cell proliferation significantly in a dose and time-dependent manner in SUM149 cells. Cells were treated with 0, 500, 1000, 2500, and 5000 U/ml of IFN-α for 24 and 48 hours. At each time point, the IFN-α was removed, the cells rinsed with PBS, and cell proliferation was evaluated (* P < 0.05, ** P < 0.01, (*** P < 0.001). The reduction in cell proliferation induced by treatment with IFN-α is an IBC-specific response. The 2 non-IBC cell lines (A) MDA-MB-231 and (B) BT474 did not demonstrate a significant decrease in cell proliferation with IFN-α treatment (* P < 0.05, ** P < 0.01, *** P < 0.001).
Figure 5
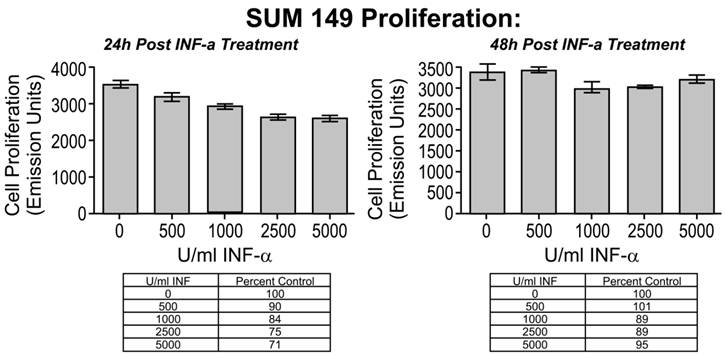
IFN- α response abrogated with antibody block. (A) SUM149 cells were treated for 24 hours with increased doses of IFN-α; cell proliferation decreased in a dose-dependent manner. (B) IFN-α complexed to IFN-α specific antibody abrogated the dose-dependent decrease in cell proliferation. For IFN-α block experiments, each treatment was pre-incubated for 15 minutes with 1 μg/ml IFN-α specific antibody (* P < 0.05, ** P < 0.01, *** P < 0.001).
Figure 6
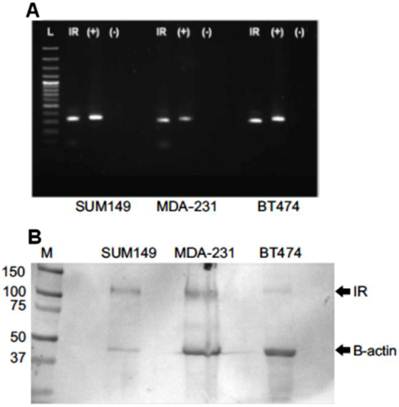
(A) Reverse transcription polymerase chain reaction (RT-PCR) and (B) Western blot for human IFN-α Receptor. (A) RT-PCR showed that the IFN- α receptor was basally expressed in untreated cultured SUM149, MDA-MB-231, and BT474 cells. L: DNA ladder, IR: IFN-α receptor, (+): GAPDH controls, (-): no template negative controls. (B) Western blot analysis indicating human IFN-α receptor protein expressed in the membrane fraction of SUM149, MDA-MB-231, and BT474 cell lines. M: Molecular weight marker, IR: Interferon-alpha receptor type I. β-actin served as controls for SDS-PAGE protein loading and for complete immunoblot transfer.
PCR-based analysis of HMTV sequences in SUM149
The results of our PCR-based analysis to detect the presence of HMTV sequences are shown in Figure 7. PCR analysis of genomic DNA (Figure 7A) and RT-PCR analysis (Figure 7B) of 4 primer sets for the HMTV env/LTR and late LTR regions showed that each of these viral elements were not detected in the SUM149 cell line. In addition, our Southern blot analysis using a specific probe to the env region of HMTV did not detect the presence of HMTV integrated in the SUM149 genome (Figure 8), consistent with our PCR findings. Although we were able clone and sequence PCR amplicons generated by our analyses, none of these fragments revealed homology to HMTV (data not shown). Our findings do not indicate the presence of putative HMTV in SUM149 cells.
Figure 7
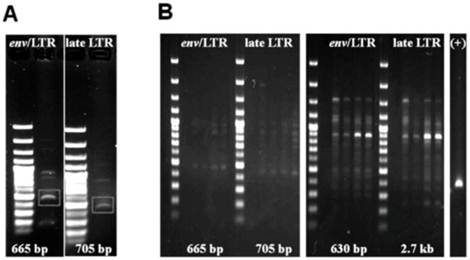
PCR products from SUM149 genomic DNA. Agarose gel electrophoresis of PCR amplicons from genomic DNA (A) and RT-PCR amplicons from RNA (B) using 4 primer sets (see materials and methods for sequences) for the HMTV env/LTR and late LTR regions. Expected product sizes if HMTV sequences were present are shown at the bottom of each gel. A. PCR analysis resulted in nonspecific amplicons from chromosomes 7 and 17. The white box highlights bands of interest that were cloned and sequenced. B. RT-PCR using several primer pairs over an annealing temperature gradient produced nonspecific amplicons, as compared with the positive controls (+) GAPDH. Molecular weight marker was 47kDa.
Figure 8
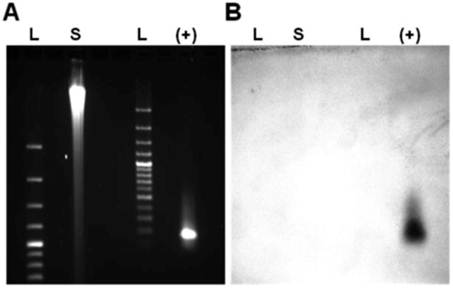
Southern blot analysis for HMTV sequences in SUM149 genome. Southern blot analysis indicated the env region of HMTV was not present in the SUM149 genome. (A) Ethidium bromide visualization of BamHI-digested and gel electrophoresed SUM149 genomic DNA and a synthetic 172 base-pair HMTV fragment. (B) Southern blot probed with biotinylated HMTV env probe. L: DNA ladder, S: 10 μg digested SUM149 genomic DNA, (+): HMTV controls.
DISCUSSION
Our experimental analysis of IBC cell lines SUM149 and SUM190, revealed 2 SNPs in the RNase L gene with known association to prostate carcinogenesis, particularly those of possible viral etiology. The consistent IBC homozygous variants 462R and 541E were infrequent or absent in non-IBC cell lines respectively suggesting these variants may represent novel risk alleles for IBC onset. This finding warrants investigation in patient samples. In an effort to determine if these SNPs could portend a genetic predisposition to IBC, we first performed an analysis of RNase L transcript levels between IBC and non-IBC breast tissue samples from publicly available data. Within the stroma of breast tissue, there was no significant difference in the gene expression of these RNase L SNPs in IBC and non-IBC tissue samples (Figure 3). However, the expected differences in prevalence, if there are any, should be in the somatic tissues, such as breast parenchyma. This data suggests that altered activity of RNase L is not likely due to altered abundance.
In 2012, Jin et al. 33 reported using two sequence homology-based computational tools [Sort Intolerant from Tolerant (SIFT) and Polymorphism Phenotype (PolyPhen)] to predict the functional contributions of several non-synonymous RNase L variants. Their analysis suggested the R462Q and E541D variants are predicted to be 'tolerated' changes, however functional studies on these SNPs are yet to be conducted, particularly in the context of prior viral infection. Further, despite the prediction of a tolerated change, variants at RNase L amino acid residues 462 and 541 have been definitively associated with altered cancer predisposition.
Notably, Next Generation Sequencing would yield the most appropriate dataset for this comparison, but such datasets are not currently available. A significant confounding factor for a study of this nature is the paucity of archived IBC samples, owing to its rare clinical presentation and particular difficulties with biospecimen collection of this tumor type, which typically lacks a mass lesion and is generally treated with systemic therapy prior to surgery 13. Further validation with patient samples is required to evaluate the frequency of RNase L variants in IBC, and whether they are indeed a biomarker of genetic risk for IBC. Of note, the sample detection could be through patient cheek swaps, since the polymorphism would likely affect all somatic tissues. In turn, tumor isolation may not be necessary for SNP detection in IBC patients. Our findings suggest that the 541G variant may serve as a susceptibility factor in the development of IBC, but larger sample sizes are needed to better assess the role of 462G, which may potentially serve as an IBC biomarker.
In contrast to our negative findings for HMTV in IBC DNA, our finding of a selective response to IFN-α treatment may indicate the potential to respond to viral infection in IBC, as the 2-5A pathway is a key mediator in the innate response to viral infection 37. Additionally, RNase L 462R and 541 have normal enzymatic functionality (both 462 and 541 variants are in the RNase L protein kinase domain), and may, in turn, decrease the likelihood of viral infection 31.
It is not without precedent that cell lines have been found to have a constitutive viral load. Pattillo et al. have shown that the cervical cancer cell line CaSKI has an average viral load of 600 particles per cell 43. However, a constitutive viral load has yet to be demonstrated within IBC cell lines.
Of note, Reuben et al. recently suggested that Epstein-Barr virus (EBV) may also play a potential role in the pathogenesis of IBC 44. They demonstrated that 20% of IBC patients have been exposed to EBV, as determined by the detection of EBV-specific immunoglobulin G (IgG) antibody from peripheral blood 44. They also demonstrated that a large portion of IBC patients have reactivated EBV infection. However, no causal relationship between EBV and IBC has been proven, so the role of EBV in IBC is yet to be determined, despite the more clearly defined role of EBV in carcinogenesis of diseases such as nasopharyngeal carcinoma and Burkitt's lymphoma 45.
Concomitantly, in our endeavor to detect the presence of the HMTV virus, we did not detect HMTV env sequences, by either RT-PCR or Southern blot analysis, in SUM149 cells. In contrast, Pogo et al. recently reported an increased detection of HMTV in IBC samples and increased expression of HMTV envelope (Env) and capsid (Ca) proteins in 10 primary cultures of human breast cancer containing HMTV sequences (MSSM) 46. These cells were derived from discarded ascitic fluids or pleural effusions obtained from patients with metastatic breast cancer 46.
Pogo et al. also reported that, by using nested priming PCR and Southern blot techniques, they detected the presence of MMTV-like env sequences in 71.5% of the 67 human tissue samples evaluated 46-48. Although those studies by Pogo et al. are thought-provoking, a causal relationship has yet to be definitively established, given the conflicting nature of the reports currently available on the prevalence of MMTV-like sequences within IBC patient specimens 49,50. In our study, both our PCR and Southern blot analysis failed to detect HMTV-like env sequences in the SUM149 cell line—despite the lower limits of detection in our Southern blot assay being in the femtogram level 51.
Although the findings of Pogo et al. conflicted with ours in that they found HMTV in tumors from IBC patients, some marked differences between the 2 models could explain this discrepancy 25. In our study, we used authenticated SUM149 cells that were derived from a primary inflammatory ductal carcinoma of the breast and established as an immortalized cell line that is well characterized 27-29. To our knowledge, no other group has investigated the presence of putative viral HMTV elements in this cell line, so our results provide novel insights, as SUM149 is the most widely used in vitro model for IBC.
Even though SUM149 cells serve as an excellent IBC model, the dynamics of disease progression may differ in this established cell line, as compared with the primary tumors evaluated by Pogo et al. Given the rarity of IBC clinical specimens, we were not able to examine tumors for viral sequences in our in vitro study. Our power calculation of the minimum sample size required to have 80% power to detect at least a 2-fold increase in risk between IBC and non-IBC cancer is 356 samples of each cancer (IBC and non-IBC) with 160 healthy (control) samples. Given that few institutions would be likely to have 356 IBC banked specimens or access to 356 IBC patients currently being followed in the clinic who might participate in the evaluation of the prevalence of these RNase L SNPs, validation of these SNPs becomes problematic. Moreover, clinical biomarker validation studies with fewer than the calculated number of patients may be misleading or even futile 52.
However, given the large number of IBC specimens required to establish whether these SNPs correlate with IBC, this study would be best performed using either multi-institutional datasets or in the context of a clinical trial's biospecimen collection. One such approach would be genome wide association studies (GWAS) of relevant datasets to query the prevalence of RNase L SNPs in IBC; unfortunately, no such dataset exists at this time.
However, since SUM149 is the most widely studied IBC cell line model, our finding that it may be discordant with regard to the presence of the HMTV genome in a cohort of IBC primary tumors calls into question its appropriateness as a model for IBC, provided that the findings of Pogo et al. are validated. Nonetheless, in the absence of independent confirmation of Pogo et al.'s findings that HMTV is prevalent in IBC, SUM149 remains the commercially available in vitro model of choice for IBC.
Alternatively, our in vitro study has identified 2 SNPs, specific genotypic variants of which identify important possible genetic risk determinants for IBC. Future studies investigating the genotypic frequency of these SNPs within human IBC tumors are warranted to validate our in vitro findings.
Acknowledgements
Financial support: seed funds from the Arizona Cancer Center and the Department of Surgery, University of Arizona. NIH P30CA014089 supports the USC Norris Comprehensive Cancer Center. NIH grant P30CA023074 supports the Arizona Cancer Center.
This study was presented in part at the 2011 American Society of Clinical Oncology Breast Cancer Symposium, San Francisco, CA.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Gonzalez-Angulo AM, Hennessy BT, Broglio K. et al. Trends for inflammatory breast cancer: is survival improving? Oncologist. 2007;12:904-12
2. Bristol IJ, Woodward WA, Strom EA. et al. Locoregional treatment outcomes after multimodality management of inflammatory breast cancer. Int J Radiat Oncol Biol Phys. 2008;72:474-84
3. Charafe-Jauffret E, Tarpin C, Viens P, Bertucci F. Defining the molecular biology of inflammatory breast cancer. Semin Oncol. 2008;35:41-50
4. Hance KW, Anderson WF, Devesa SS, Young HA, Levine PH. Trends in inflammatory breast carcinoma incidence and survival: the surveillance, epidemiology, and end results program at the National Cancer Institute. J Natl Cancer Inst. 2005;97:966-75
5. Dirix LY, Van Dam P, Prove A, Vermeulen PB. Inflammatory breast cancer: current understanding. Curr Opin Oncol. 2006;18:563-71
6. Singletary SE, Cristofanilli M. Defining the clinical diagnosis of inflammatory breast cancer. Semin Oncol. 2008;35:7-10
7. Dawood S, Merajver SD, Viens P. et al. International expert panel on inflammatory breast cancer: consensus statement for standardized diagnosis and treatment. Ann Oncol. 2011;22:515-23
8. Lerebours F, Bieche I, Lidereau R. Update on inflammatory breast cancer. Breast cancer research: BCR. 2005;7:52-8
9. Tai P, Yu E, Shiels R. et al. Short- and long-term cause-specific survival of patients with inflammatory breast cancer. BMC Cancer. 2005;5:137
10. Cristofanilli M. Novel targeted therapies in inflammatory breast cancer. Cancer. 2010;116:2837-9
11. Dushkin H, Cristofanilli M. Inflammatory breast cancer. J Natl Compr Canc Netw. 2011;9:233-40
12. Ellis GK, Barlow WE, Gralow JR. et al. Phase III comparison of standard doxorubicin and cyclophosphamide versus weekly doxorubicin and daily oral cyclophosphamide plus granulocyte colony-stimulating factor as neoadjuvant therapy for inflammatory and locally advanced breast cancer: SWOG 0012. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2011;29:1014-21
13. Smoot RL, Koch CA, Degnim AC. et al. A single-center experience with inflammatory breast cancer, 1985-2003. Arch Surg. 2006;141:567-72
14. Taghian A, El-Ghamry MN, Merajver SD. Inflammatory breast cancer: Clinical features and treatment. Waltham, MA: Uptodate. 2011
15. Robertson FM, Bondy M, Yang W. et al. Inflammatory breast cancer: the disease, the biology, the treatment. CA Cancer J Clin. 2010;60:351-75
16. Gonzalez-Angulo AM, Sneige N, Buzdar AU. et al. p53 expression as a prognostic marker in inflammatory breast cancer. Clin Cancer Res. 2004;10:6215-21
17. Houchens NW, Merajver SD. Molecular determinants of the inflammatory breast cancer phenotype. Oncology (Williston Park). 2008;22:1556-61
18. Pogo BG, Melana SM, Holland JF. et al. Sequences homologous to the mouse mammary tumor virus env gene in human breast carcinoma correlate with overexpression of laminin receptor. Clin Cancer Res. 1999;5:2108-11
19. Fantozzi A, Christofori G. Mouse models of breast cancer metastasis. Breast cancer research: BCR. 2006;8:212
20. Holland JF, Pogo BG. Mouse mammary tumor virus-like viral infection and human breast cancer. Clin Cancer Res. 2004;10:5647-9
21. Fernandez-Cobo M, Melana SM, Holland JF, Pogo BG. Transcription profile of a human breast cancer cell line expressing MMTV-like sequences. Infect Agent Cancer. 2006;1:7
22. Bindra A, Muradrasoli S, Kisekka R, Nordgren H, Warnberg F, Blomberg J. Search for DNA of exogenous mouse mammary tumor virus-related virus in human breast cancer samples. The Journal of general virology. 2007;88:1806-9
23. Mok MT, Lawson JS, Iacopetta BJ, Whitaker NJ. Mouse mammary tumor virus-like env sequences in human breast cancer. Int J Cancer. 2008;122:2864-70
24. Etkind PR, Stewart AF, Wiernik PH. Mouse mammary tumor virus (MMTV)-like DNA sequences in the breast tumors of father, mother, and daughter. Infect Agent Cancer. 2008;3:2
25. Pogo BG, Melana SM, Moran H, Holland JF. Presence of MMTV-like env gene sequences in human breast cancer. Breast Cancer Res Treat. 2011;125:295-7
26. Pogo BG, Holland JF, Levine PH. Human mammary tumor virus in inflammatory breast cancer. Cancer. 2010;116:2741-4
27. Ethier SP, Mahacek ML, Gullick WJ, Frank TS, Weber BL. Differential isolation of normal luminal mammary epithelial cells and breast cancer cells from primary and metastatic sites using selective media. Cancer Res. 1993;53:627-35
28. Forozan F, Veldman R, Ammerman CA. et al. Molecular cytogenetic analysis of 11 new breast cancer cell lines. Br J Cancer. 1999;81:1328-34
29. Hoffmeyer MR, Wall KM, Dharmawardhane SF. In vitro analysis of the invasive phenotype of SUM 149, an inflammatory breast cancer cell line. Cancer cell international. 2005;5:11
30. Rokman A, Ikonen T, Seppala EH. et al. Germline alterations of the RNASEL gene, a candidate HPC1 gene at 1q25, in patients and families with prostate cancer. Am J Hum Genet. 2002;70:1299-304
31. Shook SJ, Beuten J, Torkko KC. et al. Association of RNASEL variants with prostate cancer risk in Hispanic Caucasians and African Americans. Clin Cancer Res. 2007;13:5959-64
32. Urisman A, Molinaro RJ, Fischer N. et al. Identification of a novel Gammaretrovirus in prostate tumors of patients homozygous for R462Q RNASEL variant. PLoS Pathog. 2006;2:e25
33. Jin W, Wu DD, Zhang X, Irwin DM, Zhang YP. Positive Selection on the Gene RNASEL: Correlation between Patterns of Evolution and Function. Molecular biology and evolution. 2012
34. De la Vega FM, Lazaruk KD, Rhodes MD, Wenz MH. Assessment of two flexible and compatible SNP genotyping platforms: TaqMan SNP Genotyping Assays and the SNPlex Genotyping System. Mutation research. 2005;573:111-35
35. Boersma BJ, Reimers M, Yi M. et al. A stromal gene signature associated with inflammatory breast cancer. Int J Cancer. 2008;122:1324-32
36. Sen GC. Viruses and interferons. Annu Rev Microbiol. 2001;55:255-81
37. Samuel CE. Antiviral actions of interferons. Clin Microbiol Rev. 2001;14:778-809
38. Silverman RH. A scientific journey through the 2-5A/RNase L system. Cytokine & growth factor reviews. 2007;18:381-8
39. Silverman RH. Viral encounters with 2',5'-oligoadenylate synthetase and RNase L during the interferon antiviral response. J Virol. 2007;81:12720-9
40. Pfeiffer RM, Gail MH. Sample size calculations for population- and family-based case-control association studies on marker genotypes. Genet Epidemiol. 2003;25:136-48
41. Kaiser J. Chronic fatigue syndrome. Studies point to possible contamination in XMRV findings. Science. 2011;331:17
42. Smith RA. Contamination of clinical specimens with MLV-encoding nucleic acids: implications for XMRV and other candidate human retroviruses. Retrovirology. 2010;7:112
43. Pattillo RA, Hussa RO, Story MT, Ruckert AC, Shalaby MR, Mattingly RF. Tumor antigen and human chorionic gonadotropin in CaSki cells: a new epidermoid cervical cancer cell line. Science. 1977;196:1456-8
44. Reuben J. Epstein-Barr Virus (EBV) in Patients with Inflammatory Breast Cancer: A Potential Role in Pathogenesis of the Disease. First International Inflammatory Breast Cancer Conference. Houston, Texas: Wiley-Blackwell. 2008
45. Kaneda A, Matsusaka K, Aburatani H, Fukayama M. Epstein-barr virus infection as an epigenetic driver of tumorigenesis. Cancer Res. 2012;72:3445-50
46. Pogo BG. Human Mammary Tumor Virus and IBC. First International Inflammatory Breast Cancer Conference. 2008 Houston, Texas: Blackwell-Wiley 2008
47. Melana SM, Nepomnaschy I, Hasa J, Djougarian A, Holland JF, Pogo BG. Detection of human mammary tumor virus proteins in human breast cancer cells. J Virol Methods. 2010;163:157-61
48. Levine PH, Young H. A, Mark L, Rojowsky H, Holland J. F, Pogo B. G.. Increased detection of breast cancer virus sequences in inflammatory breast cancer. Advances in Tumor Virology. 2009;1:3-7
49. Stewart A. MMTV-related env sequences in human breast tumor. Int J Cancer. 2003;106:138
50. Park DJ, Southey MC, Giles GG, Hopper JL. No evidence of MMTV-like env sequences in specimens from the Australian Breast Cancer Family Study. Breast Cancer Res Treat. 2011;125:229-35
51. Murakami S, Mizobuchi M, Nakashiro Y, Doi T, Hato N, Yanagihara N. Bell palsy and herpes simplex virus: identification of viral DNA in endoneurial fluid and muscle. Ann Intern Med. 1996;124:27-30
52. Gail MH, Pfeiffer RM, Wheeler W, Pee D. Probability of detecting disease-associated single nucleotide polymorphisms in case-control genome-wide association studies. Biostatistics. 2008;9:201-15
Author contact
![]() Corresponding author: Julie E. Lang, email: Julie.Langusc.edu. Address: 1510 San Pablo Street, Suite 412, Los Angeles, CA 90033 FAX: 323-865-3539 PHONE: 323-422-5772.
Corresponding author: Julie E. Lang, email: Julie.Langusc.edu. Address: 1510 San Pablo Street, Suite 412, Los Angeles, CA 90033 FAX: 323-865-3539 PHONE: 323-422-5772.
Citation styles
APA
Nokes, B.T., Cunliffe, H.E., LaFleur, B., Mount, D.W., Livingston, R.B., Futscher, B.W., Lang, J.E. (2013). In Vitro Assessment of the Inflammatory Breast Cancer Cell Line SUM 149: Discovery of 2 Single Nucleotide Polymorphisms in the RNase L Gene. Journal of Cancer, 4(2), 104-116. https://doi.org/10.7150/jca.5002.
ACS
Nokes, B.T.; Cunliffe, H.E.; LaFleur, B.; Mount, D.W.; Livingston, R.B.; Futscher, B.W.; Lang, J.E. In Vitro Assessment of the Inflammatory Breast Cancer Cell Line SUM 149: Discovery of 2 Single Nucleotide Polymorphisms in the RNase L Gene. J. Cancer 2013, 4 (2), 104-116. DOI: 10.7150/jca.5002.
NLM
Nokes BT, Cunliffe HE, LaFleur B, Mount DW, Livingston RB, Futscher BW, Lang JE. In Vitro Assessment of the Inflammatory Breast Cancer Cell Line SUM 149: Discovery of 2 Single Nucleotide Polymorphisms in the RNase L Gene. J Cancer 2013; 4(2):104-116. doi:10.7150/jca.5002. https://www.jcancer.org/v04p0104.htm
CSE
Nokes BT, Cunliffe HE, LaFleur B, Mount DW, Livingston RB, Futscher BW, Lang JE. 2013. In Vitro Assessment of the Inflammatory Breast Cancer Cell Line SUM 149: Discovery of 2 Single Nucleotide Polymorphisms in the RNase L Gene. J Cancer. 4(2):104-116.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) License. See http://ivyspring.com/terms for full terms and conditions.