Journal of Cancer
ISSN: 1837-9664
3.2
Impact Factor
ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2013; 4(1):84-95. doi:10.7150/jca.5482 This issue Cite
Review
Tumor-Infiltrating Immune Cells Promoting Tumor Invasion and Metastasis: Existing Theories
Yan-gao Man1,2 ![]() , Alexander Stojadinovic3, Jeffrey Mason4, Itzhak Avital5, Anton Bilchik6, Bjoern Bruecher7, Mladjan Protic8, Aviram Nissan9, Mina Izadjoo1, Xichen Zhang2, Anahid Jewett10
, Alexander Stojadinovic3, Jeffrey Mason4, Itzhak Avital5, Anton Bilchik6, Bjoern Bruecher7, Mladjan Protic8, Aviram Nissan9, Mina Izadjoo1, Xichen Zhang2, Anahid Jewett10 ![]()
1. Diagnostic and Translational Research Center, Henry Jackson Foundation, Gaithersburg, MD, USA;
2. College of Animal Science and Veterinary Medicine, Jilin University, Changchun, Jilin, China;
3. Surgical Oncology, Walter Reed National Military Medical Center, and Uniformed Services University of the Health Sciences, Bethesda, MD, USA;
4. Veterans Affair Medical Center, Washington, DC, USA;
5. Bon Secours National Cancer Institute (BSNCI), Richmond VA, USA;
6. John Wayne Cancer Institute; California Oncology Research Institute; and, David Geffen School of Medicine, University of California, Los Angeles, USA;
7. Theodor-Billroth-Academy®, Munich, Germany;
8. Clinic of Abdominal, Endocrine, and Transplantation Surgery, Clinical Center of Vojvodina, University of Novi Sad - Medical Faculty, Novi Sad, Serbia;
9. The Surgical Oncology Laboratory, Department of Surgery, Hadassah-Hebrew University Medical Center, Mount Scopus, Jerusalem, Israel;
10. Division of Oral Biology and Medicine, Jonsson Comprehensive Cancer Center, UCLA School of Dentistry, Los Angeles, CA, USA.
Received 2012-11-4; Accepted 2012-12-20; Published 2013-1-5
Citation:
Man Yg, Stojadinovic A, Mason J, Avital I, Bilchik A, Bruecher B, Protic M, Nissan A, Izadjoo M, Zhang X, Jewett A. Tumor-Infiltrating Immune Cells Promoting Tumor Invasion and Metastasis: Existing Theories. J Cancer 2013; 4(1):84-95. doi:10.7150/jca.5482. https://www.jcancer.org/v04p0084.htm
Other stylesAbstract
It is a commonly held belief that infiltration of immune cells into tumor tissues and direct physical contact between tumor cells and infiltrated immune cells is associated with physical destructions of the tumor cells, reduction of the tumor burden, and improved clinical prognosis. An increasing number of studies, however, have suggested that aberrant infiltration of immune cells into tumor or normal tissues may promote tumor progression, invasion, and metastasis. Neither the primary reason for these contradictory observations, nor the mechanism for the reported diverse impact of tumor-infiltrating immune cells has been elucidated, making it difficult to judge the clinical implications of infiltration of immune cells within tumor tissues. This mini-review presents several existing hypotheses and models that favor the promoting impact of tumor-infiltrating immune cells on tumor invasion and metastasis, and also analyzes their strength and weakness.
Keywords: immune cell, tumor progression, invasion, metastasis
Background
Cancer has been detected in all epithelium-derived tissues of the human body. Carcinogenesis in different organs is believed to share a similar process, i.e, sequential progression from normal to hyperplasia, to in situ, and then, to invasive or metastatic cancer [1-4]. A vast majority of in situ cancer can be cured by surgical resection alone, while invasive and metastatic cancer accounts for over 90% of cancer-related mortality [5-8]. The significant difference in clinical prognosis between in situ and invasive or metastatic cancer results predominantly from the presence or absence of the surrounding basement membrane (BM). All normal or pre-invasive tumor epithelia are normally devoid of lymphatic ducts and blood vessels and are also physically segregated from vascular structures within the stroma by the BM. The BM consists of mainly type IV collagen, laminins, and other molecules that form a continuous sheet (more commonly called the tumor capsule), surrounding the epithelial cells [9-12]. In human breast, prostate, and major salivary glands, the capsule is further reinforced by a single layer of elongated cells, which are named “myoepithelial” cells in the breast and salivary glands; and “basal cells” in the prostate. The basal or myoepithelial cell layer lies between the epithelial cells and the BM. In the gastrointestinal tract, the normal mucosa and in situ cancer are further separated from the submucosa by the muscularis mucosa, a dense band comprised of two layers of smooth muscle cells [13]. Due to these structural relationships, the disruption of the tumor capsule and its associated physical barriers is an absolute pre-requisite for tumor cell invasion or metastasis.
It is a commonly held belief that progression from in situ to invasive or metastatic cancer is caused by proteolytic enzymes produced by tumor cells that increase linearly in concentration with tumor progression, reaching their highest level at the in situ cancer stage. It has been proposed that these proteolytic enzymes cause degradation or disruption of the tumor capsule and allow the in situ cancer cells to migrate into the adjacent stroma or to disseminate to distant organs [14-17]. The above model of tumor invasion and metastasis is consistent with results obtained from tissue culture and animal model studies [18-20]; however, it is hard to reconcile with a number of well-established observations: (1) although a vast majority of tumor cells express high levels of proteolytic enzymes, only 10-30% of untreated in situ cancers progress to invasive or metastatic cancer [21-25]; (2) the outcomes of world-wide clinical trials with proteolytic enzyme-targeted inhibitors have yielded very disappointing results [26,27]; (3) prostate tissues from many cancer-free men harbor a DNA phenotype identical to that of invasive prostate cancer [28,29]; and (4) cancer of unknown primary site is one of the ten most frequent cancers and the 4th highest cause of cancer-related mortality, despite the lack of an identifiable primary tumor to serve as a source of metastatic tumor cells [30]. Together, these facts argue that alternative pathways may exist for tumor progression and subsequent invasion or metastasis.
Existing hypotheses of tumor infiltrating immune cells promoting tumor invasion and metastasis
A great number of studies have shown that infiltration of the immune cells into tumor tissues and direct physical contact between infiltrating immune cells and tumor cells are associated with the physical destruction of tumor cells, reduction of the tumor burden, and an improved clinical prognosis [31-36]. On the other hand, a significant and steadily increasing number of studies have shown that increased infiltration of immune cells may promote tumor progression and invasion. For example, several studies have documented that stage- and histopathologically-matched pre-invasive prostate and esophageal tumors with increased immune cell infiltration have a significantly higher frequency of subsequent progression to invasive cancer than their counterparts without aberrant immune cell infiltration [37-39]. Unfortunately, the primary reasons for these contradictory observations remain elusive, making it difficult to judge the clinical implications of the infiltration of immune cells within tumor tissues. To address these issues, numerous studies [40-47] have been conducted, and a number of hypotheses [48-55] have been presented to explore the primary impact of tumor infiltrating immune cells on associated tumor tissues. Again, the outcomes of these studies are highly contradictory and the primary impact of infiltrating immune cells on associated tumors remains elusive [56-57]. In this mini-review, we present several existing hypotheses that favor the promoting impact of tumor-infiltrating immune cells on tumor invasion and metastasis and analyze their strengths and weakness. These specific hypotheses were selected for a number of reasons, including (1) they directly address the impact of tumor infiltrating immune cells on tumor cell behavior, and (2) they appear to be applicable to multiple, or perhaps all, epithelial-derived tumors.
1. Tumor-educated macrophages (paracrine loop signaling)
The main concept of this hypothesis was introduced in 2004 [58] and was based on findings of a chemotaxis-based in vivo invasion assay and multiphoton-based intravital imaging on transgenic mice. The study shows that interactions between breast tumor cells and macrophages facilitates the migration of cancer cells into the primary tumor and also that tumor cell intravasation occurs in association with perivascular macrophages [59,60]. According to this hypothesis, “macrophages are recruited to the invasive front by expression of tumor-derived chemotactic factors and in response to the disruption of the basement membrane. At this invasive site, macrophages enhance tumor cell migration and invasion through their secretion of chemotactic and chemokinetic factors including epidermal growth factor (EGF). They promote angiogenesis by the synthesis of angiogenic factors including vascular endothelial growth factor (VEGF), and they remodel the extracellular matrix and in particular, regulate collagen fibrillogenesis. A combination of these factors provides a triple-whammy, as the more mobile and invasive tumor cells track along collagen fibers that are also anchored to blood vessels, which are fabricated at sites of invasion and through which macrophages potentiate tumor cell intravasation” [61].
The main strength of this hypothesis is that the assay system used in these studies allows direct visualization of macrophage-assisted tumor cell migration and intravasation in mammary tumors. The primary weakness of this hypothesis is that it may not truly reflect the intrinsic events in humans for four important reasons. (1). Previous studies have shown that human macrophages are significantly different from mouse macrophages not only in their relative ratio to other immune cell types, but also in their use of arginine for production of nitric oxide (NO), which is the most important component of the macrophage arsenal against intracellular pathogens [62-69]. The mouse macrophages produce large amounts of NO and L-citrulline from L-arginine via induction of the inducible form of NOS (iNOS), and also synthesize the obligatory cofactor tetrahydrobiopterin (BH4), essential for stabilization and function of the iNOS enzyme protein [63,64]. The human macrophages, however, do not have NOS activity nor do they synthesize BH4 [65-69]. (2). The human immune system is also fundamentally distinct from the mouse immune system in development, activation, and response to challenge, in both innate and adaptive immunity [70-73]. The human peripheral blood is neutrophil rich (50-70% neutrophils, 30-50% lymphocytes), whereas the mouse peripheral blood is predominantly lymphocytes (75-90% lymphocytes, 10-25% neutrophils) [70]. The putative human hemopoietic stem cells (HSC) express predominantly c-kitlow, flt-3+, whereas the mouse HSC express predominantly c-kithigh, flt-3− [71]. Similarly, the human neutrophils are a rich source of leukocyte defensins, whereas defensins are not expressed by neutrophils in mice [72-74]. (3). It has been well documented that the primary function of the human macrophages is to remove cell debris and infiltrated microorganisms after tissue injury [75-79]. It has also been documented that aberrant accumulation of macrophages often has destructive impact on their associated tissues [80]. (4). In the adult organs, only stem cells retain the potential for unlimited proliferation and multi-lineage differentiation, and, consequently, stem cells have been considered as the primary source for invasive and metastatic lesions [81-85]. The paracrine loop signaling hypothesis, however, has failed to address the role of tumor stem cells in invasion or metastasis. Collectively, these weaknesses make it difficult to determine whether, or to what extent, this hypothesis truly reflects the intrinsic events of human tumor invasion or metastasis.
2. Immune cell-based mediation
This hypothesis is based on studies using the polyoma-middle-T-antigen (PyMT) transgeneic mouse model of mammary carcinogenesis [86]. According to this hypothesis, “IL-4-expressing CD4+ T lymphocytes indirectly promote invasion and subsequent metastasis of mammary adenocarcinomas by directly regulating the phenotype and effector function of tumor-associated CD11b+Gr1-F4/80+ macrophages that in turn enhance metastasis through activation of epidermal growth factor receptor signaling in malignant mammary epithelial cells”. More recently, the same concept and similar pathways, have been extended to lymphocytes and their subtypes [87-90]. This includes the recruitment of macrophages through expression of colony-stimulating factor 1 (CSF1) by mouse mammary epithelium [90].
The main strength of this hypothesis is that it presents a broader view of the potential impact of interactions among different immune cell types on tumor progression. The primary weakness of this hypothesis is that it is based on studies using mouse models and thus raises the same issues with applicability to human tumor tissues as discussed above. In addition, the model focuses predominately on pathways that facilitate late-stage promotion of tumor progression to metastasis.
3. Cancer cell-leukocyte fusion
This hypothesis is the extension of a century-old theory introduced by a German pathologist, Aichel O, in 1911 [91]. Based on this hypothesis, macrophages ingest tumor cells leading to the fusion of genetic materials from the two cell types, resulting in the creation of a hybrid phenotype that exhibits chemotactic migration in vitro toward fibronectin and shows high frequencies of metastasis when implanted in mice [92-97].
The main strength of this hypothesis is its relevance to human carcinogenesis as both cancer cell- and macrophage-specific molecules are detectable in a subset of human clinical tumor samples [92-97]. The mechanism upon which this hypothesis is based, however, is not likely to represent a major route of metastatic cancer for a simple reason: if the fusion of macrophages and cancer cells is indeed the precursor of metastatic cancer, all metastatic cancer cells from different organs and histolological origins should share the same or similar morphology, specifically a giant cell population with polyploidy. However, it is well established that a majority of metastatic cancer cells are morphological and immunohistochemically similar to their primary tumors, which are highly heterogeneous in morphology.
4. Regulatory T cells (Treg) induced immune suppression
This hypothesis is based on both clinical observations of ovarian cancer patients and laboratory findings from studies of the synegeneic ID8 ovarian cancer model [98,99]. According to this hypothesis, “recruitment of Treg cells to the tumor supports disease progression through a dual mechanism: (1) the canonical subversion of antitumor immunity, and (2) through the establishment of a proangiogenic reprogramming of the tumor microenvironment”. The authors of this hypothesis believe that the recruitment of regulatory T cells to the tumors are mediated primarily through the CCL28-CCR10 interaction [99].
The main strength of this hypothesis is that it is based on both clinical and laboratory data and that Treg-cell induced immune suppression has been well documented in multiple types of human cancer and diseases [100-104]. The main weakness of the hypothesis is that CCL28 is not a widely distributed molecule; consequently, the applicability of this hypothesis to other tumor types has yet to be established. In addition, this hypothesis fails to address the impact of immune cells on tumor stem cells, which are now believed to serve as the “seeds” for both invasive, metastatic, and recurrent cancer.
5. Monocyte-mediated protection against natural killer cell lysis of cancer stem cells
This recently introduced hypothesis based on the findings that “increased NK cell cytotoxicity was seen when they were cultured with primary oral squamous carcinoma stem cells (OSCSCs) and Glioblastoma (GBM) stem cells and not with their more differentiated counterparts. In addition, human embryonic stem cells (hESCs), human mesenchymal Stem Cells (hMSCs), and human dental pulp stem cells (hDPSCs) and human induced pluripotent stem cells (hiPSCs) were significantly more susceptible to NK cell-mediated cytotoxicity than their differentiated counterparts or parental cells from which they were derived, suggesting that NK cells were preferentially targeting and lysing stem cells and not their differentiated counterparts. It was also found that inhibition of differentiation or reversion of cells to a less-differentiated phenotype by blocking NF-κB or targeted knockdown of COX2, significantly augmented NK cell functions”. In addition, it was also found that “total population of monocytes and those depleted of CD16+ subsets were able to substantially prevent NK cell-mediated lysis of OSCSCs, MSCs and DPSCs” [105]. Furthermore, it was suggested that NK cells played a significant role in differentiation of the cells by providing critical signals via secreted cytokines as well as direct cell-cell contact after the induction of split anergy which conditioned NK cells to lose cytotoxicity and gain the ability to secrete cytokines. To be conditioned to drive differentiation, NK cells had to first receive signals through their key surface receptors either from healthy stem cells or those which had been transformed. In addition, NK cells by targeting other inflammatory cells or fibroblasts in the tumor microenvironment may become conditioned to lose cytotoxicity and gain cytokine producing phenotype before they can aid in differentiation of stem cells. These alterations in NK cell effector function could ultimately aid in driving differentiation of a population of surviving healthy as well as transformed stem cells. In cancer patients since the majority of NK cells have lost cytotoxic activity, they may eventually contribute rather than halt the progression of cancer by allowing the growth and expansion of the pool of cancer stem cells.
The main strength of this hypothesis is that it provides a general mechanism how immune cells may behave in inflammatory microenvironment for the ultimate goal of tissue regeneration and the resolution of inflammation. More importantly, this hypothesis provides a novel concept and approach to study the link between stem cells and carcinogenesis and cancer progression. Indeed, many correlates to this hypothesis has been found in the clinical setting, such as substantially decreased levels of cytotoxicity in NK cells, or increased modulation of NK cell surface antigens in cancer patients [106-114]. The weakness of this hypothesis may be that it is primarily based on findings from human cell cultures, and animal studies, and may not have taken the structural features of certain types of tumors into the consideration: the stem cell population in normal or pre-invasive cancer tissues are normally segregated from the immune cells by a dense fibrous epithelial capsule in certain tumors [9-12]. However, as noted below once the tumor capsule is disrupted conditioned NK cells may get access to drive differentiation of stem cells. On the other hand, unconditioned, cytotoxic NK cells should aid in the elimination of cancer stem cells [115-118]. Disruption of the extracellular matrix components in the tumor capsule may also be achieved via increased enzymatic digestion since conditioned NK cells and regulatory T cells are likely to secrete increased levels of degrading enzymes such as MMPs. Indeed, the presence of intraepithelial lymphocytes in the healthy and diseased gut mucosa is a good indication that CD8+ lymphocytes have access to both normal and transformed epithelial microenvironment and that their function may be important in the differentiation and maintenance of tissue integrity and repair. The events described in these studies share many common features with those described below and as such they may be complementary to our in vivo morphological observations in humans.
6. Aberrant lymphocyte infiltration-induced focal capsule disruptions
Our research group has attempted to identify more effective approaches to assess the intrinsic impact of tumor-infiltrating immune cells on tumor invasion and metastasis. In our recent studies, we compared the frequency of lymphocyte infiltration in stage- and morphology-matched human breast and prostate tumors with and without focally disrupted capsules. Of 191 breast tumor nests with focally disrupted capsules, 186 (97%) had distinct lymphocyte infiltration, compared to 46 (22%) in 207 morphologically similar counterparts with non-disrupted capsules [119]. Similarly, distinct lymphocyte infiltration was seen in 183 (91%) of 201 prostate tumor nests with focally disrupted capsules, compared to 67 (33%) in 201 morphologically similar counterparts with non-disrupted capsules [120]. Subsequent studies revealed that residual cells within focally disrupted myoepithelial or basal cell layers had a significantly lower expression of tumor suppressor and cell proliferation-related proteins, but showed a significantly higher frequency of degeneration-related changes than their morphologically similar counterparts located in regions devoid of focal disruptions [121-123]. Using double immunohistochemistry to simultaneously elucidate the BM and associated basal cell layers, all the BM-overlying focally-disrupted basal cell layers demonstrated either the presence of correlated disruptions (76 of 89, 85%) or significant attenuation of immunostaining intensity (13 of 89, 15%) [124]. Correlated BM and myoepithelial cell layer alterations were also seen in human breast tissues. In contrast to the basal or myoepithelial cells, tumor cells overlying focally disrupted capsules had a significantly higher level of proliferation and greater expression of tumor stem cell- and growth factor-related genes than their adjacent counterparts distant from the disruptions [125-131].
These findings suggest that focal basal or myoepithelial cell degeneration, aberrant lymphocyte infiltration, and capsule disruptions are likely to be correlated events that contribute to tumor progression and invasion. These findings have led to a novel hypothesis that tumor invasion or metastasis is triggered by focal capsule degeneration-induced lymphocyte infiltration that causes physical disruptions within the capsule, which selectively favors proliferation and dissemination of overlying tumor stem cells [121,122]. Based on this hypothesis, aberrant lymphocyte infiltration promotes capsule disruptions and tumor invasion through the following pathways. (1). Myoepithelial and basal cells belong to a self-renewal population that must constantly undergo both proliferation and differentiation to replace aged or injured basal cells [132-134]. Both internal and external insults, such as a predisposition of genetic defects, exposure to chemicals or radiation, and chronic inflammation, may through chronic or acute mechanisms, damage the normal stem cells in these layers, resulting in a “senescent” cell population that is prone to degeneration. (2). Degradation products of degenerated basal and myoepithelial cells, or diffusible molecules from the overlying epithelial cells, can function as self-epitopes to attract the trafficking and infiltration of immune cells into the affected sites. (3). The direct physical contact of immune cells with degenerated basal or myoepithelial cells results in the discharge of their proteolytic enzymes, leading to the physical degradation of the degenerate myoepithelial or basal cells and the surrounding local basement membrane, resulting in focal disruptions in these structures. (4). As both the basal or myoepithelial cell layers are the sole source of tumor suppressor p63 and maspin [135-138], a focal disruption could lead to several consequences: (a) a localized loss of tumor suppressors and paracrine inhibitory function would confer tumor cells with growth advantages and allow them to escape from programmed cell death [139-143], (b) a localized alteration of permeability for nutrients, growth factors, and oxygen, would selectively favor proliferation of the overlying stem cells [144-146], (c) a localized increase of lymphocyte infiltration would disrupt inter-cellular junctions and cell surface adhesion molecules, facilitating cell “budding” from the tumor core [147-150]; and (d) direct physical contact between epithelial and stromal cells would facilitate the epithelial-mesenchymal transition [151-154]. As epithelial stem cells are believed to be located overlying the basal cell layer and the BM, a focal capsule disruption at the site of genetically-altered stem cells may favor their exit from quiescence.
This new hypothesis entitled “aberrant lymphocyte infiltration-induced focal capsule disruptions” differs fundamentally from the linear model of tumor progression and the proteolytic enzyme theory of tumor invasion in the followings. (1) The direct cause of tumor capsule disruptions. According to the proteolytic enzyme theory, the disruption of the tumor capsule is directly caused by tumor cell-produced proteolytic enzymes. The new hypothesis proposes that the disruption of the tumor capsule results from focal basal cell degeneration-induced immune cell infiltration. Although the enzymes of immune cells may belong to the same proteolytic enzyme family, they are discharged only upon physical contact with aged or injured basal or myoepithelial cells. Thus, their impact is focal and independent of the tumor stage and histological grade. (2) The stage of tumor invasion. According to the enzyme theory, the level of proteolytic enzymes increases linearly with tumor progression and reaches the highest level at the in situ cancer stage, in which invasion occurs. Based on the new hypothesis, invasion can take place at any stage of carcinogenesis, requiring only that the focal capsule disruption occur at a site where the overlying epithelial layer contains tumor progenitors or stem cells. (3) The cellular origin of invasive lesions. Based on the proteolytic enzyme theory, all in situ cancer cells could contribute equally to invasive lesions. According to our hypothesis, invasive lesions are predominantly derived from tumor stem cells overlying focally disrupted tumor capsules. (4) Potential approaches for treatment, prevention, and detection of tumor invasion. According to the proteolytic enzyme theory, administration of corresponding enzyme inhibitors is the only regime for treatment and prevention of tumor invasion. Based on the new hypothesis, there are at least five such approaches: (a) to neutralize the molecules that attract immune cell infiltration, (b) to reduce the specific subtype of immune cells associated with focal capsule disruptions, (c) to develop therapeutic agents to specifically target stem cell clusters overlying focally disrupted capsules, (d) to develop therapeutic agents to stabilize the BM, and (e) to stimulate basal cell growth.
The main strength of this hypothesis is that its essential conclusions are based on morphological, immunohistochemmical, and molecular findings from multiple types of untreated human tumor tissue samples, and thus, is more likely to accurately reflect the intrinsic events of human carcinogenesis. This hypothesis has been recognized as more compatible with existing experimental evidence than the traditional “protoelytic enzyme” theory by a number of previous publications and a recently published review article from internationally recognized experts in the field [155-157]. The man weakness of this hypothesis is that it has not been able to pinpoint the specific degradation product(s) to attract lymphocyte infiltration, or the specific enzyme(s) accounting for disruptions of the tumor capsules. In addition, this hypothesis has not been able to elucidate the underlying molecular pathways that link the entire process of capsule disruptions and tumor invasion. More importantly, the clinical significance of focal capsule disruptions, immune cell infiltration near the disruptions, and “budding” cells from the disruptions has not been evaluated with clinical follow-up studies.
7. Lymphocyte-mediated cell dissemination and metastasis (the Piggy-back theory)
This new hypothesis for tumor cell dissemination and metastasis is an expansion of the hypothesis presented above (aberrant lymphocyte infiltration-induced focal capsule disruptions), which is based on new findings from our more recent studies of multiple types of human cancer, including those from breast, prostate, lung, cervix, skin, and colorectum [158-160]. These recent studies have detected almost identical frequency and pattern of focal capsule disruptions, immune cell infiltration, and cell “budding” from focally disrupted capsules, as those seen in our previous studies of breast and prostate [119-131]. More importantly, these new studies have consistently shown that aberrant tumor-infiltrating lymphocytes can trigger tumor metastasis through three correlated pathways: (a) the physical movement of infiltrated lymphocytes into the budding tumor cell nest can disrupt intercellular junctions and surface adhesion molecules, causing dissociation of some cells from the tumor core; (b) lymphocytes can conjoin with dissociated tumor cells through cell membrane fusion to form tumor-lymphocyte chimeras (TLCs); and (c) the natural ability to migrate and to cross intercellular barriers allows lymphocytes to physically drag tumor cells to remote sites and to intravasate into blood vessels or lymph ducts[158-160].
The main strength of this hypothesis is that: (1) it can reasonably explain all major events involved in metastasis, which includes dissociation from the primary site, intravasation, extravasation, migration, and colonization at distant sites, (2) it is applicable to all epithelium-derived tumors, and (3) it provides, for the first time, a morphologically defined precursor of metastatic cancer. The main weakness is that it is descriptive in nature primarily based on morphological and immunohistochemical findings on tissue sections. In addition, it has not been able to elucidate the molecular and mechanistic pathways or the specific molecules, which account for the formation of TLCs. Furthermore, the clinical significance of the presence of TLCs within the vascular structures remains to be established.
Collectively, there are three central tenants of these new hypotheses. The first is that tumor stem cells are co-localized with normal, benign, and malignant cells within capsules, while they are the only ones retaining the potential for unlimited proliferation and multi-lineage differentiation. The second is that tumor invasion or metastasis may occur at any stage of carcinogenesis if a focal capsule disruption occurs at a site where the overlying epithelium contains tumor stem cells. The third is that the interaction of lymphocytes with tumor stem cells can lead to stable adhesions between the two creating a TLC that facilitates dissemination of tumor stem cells. Based on this new hypotheses, the apparent contradiction in the role of tumor infiltrating immune cells may result primarily from the differences in tumor stages and distributions of infiltrating immune cells. As the primary tumor infiltrating immune cell types, including cytotoxic T-lymphocytes, natural killer and Mast cells, have to physically contact their targets in order to exert their cytotoxic functions [161-164], it is likely that infiltrating immune cells may be involved primarily in the physical destructions of altered epithelial capsules at the pre-invasive stage, and thus, to promote tumor invasion into the stroma. On the other hand, immune cell infiltration into the invasive tumor nests may lead to physical destruction of the tumor cells and reduction of the tumor burden. These hypotheses could reasonable explain the contradictory reports and statements regarding the impact and clinical significance of immune cell infiltration into tumor tissues. More importantly, as the disruption of the tumor capsule is an absolute prerequisite for tumor invasion and metastasis, local or systematic administration of anti-inflammatory agents to prevent immune cell infiltration-induced capsule destruction may be beneficial in preventing tumor progression. A recent report published in Lancet Oncology has revealed that regular use of Aspirin, a non-steroidal anti-inflammatory drug, reduces the long-term risk of CRC and other cancer and the risk of distant metastasis [165].
The main contents of the aberrant lymphocyte infiltration-induced focal capsule disruption andlymphocyte-mediated cell dissemination and metastasis hypotheses are depicted in Figure 1.
Conclusions
The impact of tumor-infiltrating immune cells has been subject of debate for decades. A great number of studies have shown that tumor-infiltrating immune cells are associated with the physical destruction of the tumor cells, reduction of the tumor burden, and improved clinical prognosis. On the other hand, a significant and steadily increasing number of studies have shown that increased infiltration of immune cells may promote tumor progression and invasion. In an effort to elucidate the primary impact and mechanism of tumor infiltrating immune cells on associated tumor tissues, a great number of studies have been conducted and a number of hypotheses have been presented. In this mini-review, we present several existing hypotheses that favor the promoting impact of tumor-infiltrating immune cells on tumor invasion and metastasis, and also analyze their strengths and weaknesses. These hypotheses were selected for a number of reasons, including (1) they specifically address the direct impact of tumor infiltrating immune cells on tumor behavior, and (2) they appear to be applicable to multiple or all epithelial-derived tumors. Each of these hypotheses has their individual strengths and weaknesses and are supported by laboratory findings and/or clinical data, suggesting that tumor-infiltrating immune cells may impact, directly or indirectly, associated tumors through multiple pathways and mechanisms.
Figure 1
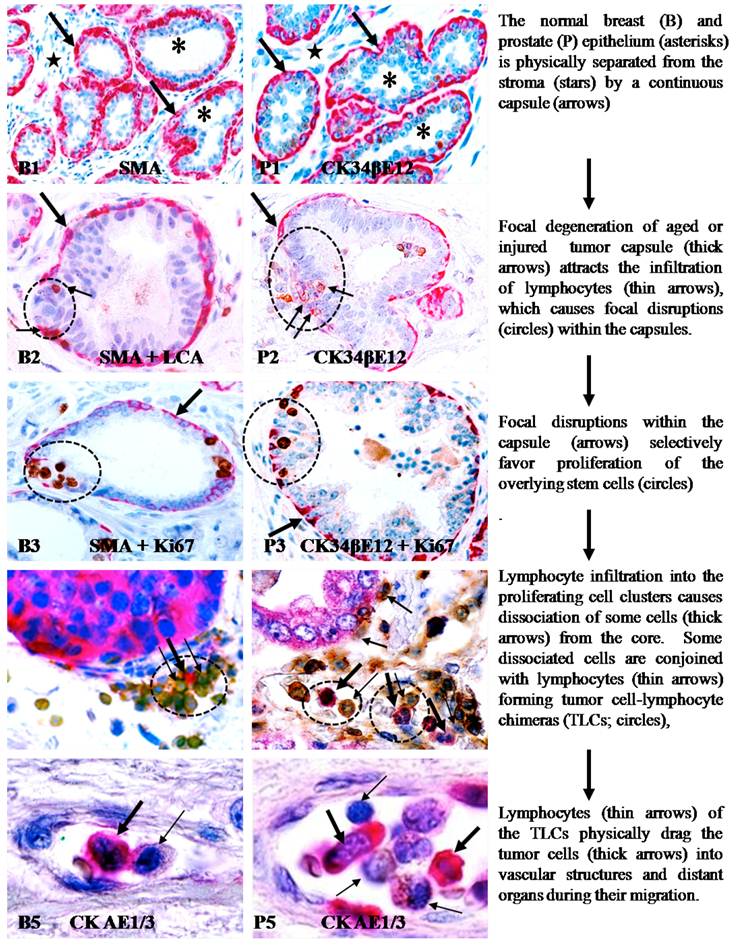
Aberrant lymphocyte infiltration-induced focal capsule disruption andlymphocyte-mediated cell dissemination and metastasis hypotheses.
Competing Interests
The authors have declared that no competing interest exists.
Disclaimer
The views expressed in this manuscript are those of the authors and do not reflect the official policy of the Department of the Army, the Department of Defense or the United States Government or the Henry Jackson Foundation.
References
1. Luebeck EG, Hazelton WD. Multistage carcinogenesis and radiation. J Radiol Prot. 2002;22(3A):A43-9
2. Moolgavkar SH, Luebeck EG. Multistage carcinogenesis and the incidence of human cancer. Genes Chromosomes Cancer. 2003Dec;38(4):302-6
3. Laconi E, Doratiotto S, Vineis P. The microenvironments of multistage carcinogenesis. Semin Cancer Biol. 2008Oct;18(5):322-9
4. Park BJ, Chiosea SI, Grandis JR. Molecular changes in the multistage pathogenesis of head and neck cancer. Cancer Biomark. 2010;9(1-6):325-39
5. Christofori G. New signals from the invasive front. Nature. 2006;441(7092):444-450
6. Edwards BK, Ward E, Kohler BA. et al. Annual report to the nation on the status of cancer, 1975-2006, featuring colorectal cancer trends and impact of interventions (risk factors, screening, and treatment) to reduce future rates. Cancer. 2010;116(3):544-73
7. Jemal A, bray F, Center MM, Ferlay J, Ward E, Forman D. Global Cancer Statistics. CA Cancer J Clin. 2011;61(2):69-90
8. Barugel ME, Vargas C, Krygier Waltier G. Metastatic colorectal cancer: recent advances in its clinical management. Expert Rev Anticancer Ther. 2009;9(12):1829-47
9. Bonkhoff H, Wernert N, Dhom G, Remberger K. Basement membranes in fetal, adult normal, hyperplastic and neoplastic human prostate. Virchows Arch A Pathol Anat Histopathol. 1991;418:375-81
10. Kosir MA, Wang W, Zukowski KL, Tromp G. Degradation of basement membrane by prostate tumor heparanase. J Surg Res. 1999;81:42-7
11. Guelstein VI, Tehypysheva TA, Ermilova VD, Liubimov AV. Myoepithelial and besement membrane antigens in benign and malignant human breast tumors. In J Cancer. 1993;53:269-77
12. Nerlich A. Morphology of basement membrane and associated matrix proteins in normal and pathological tissues. Veroff Pathol. 1995;145:1-139
13. Noffsinger A, Fenoglio-Preiser CM, Maru D, Gilinsky N. Gastrointestinal Diseases. Washington DC: American Registry of Pathology and Armed Forces Institute of Pathology. 2007:1-31
14. Goldfarb RH, Liotta LA. Proteolytic enzymes in cancer invasion and metastasis. Semin Thromb Hemost. 1986;12:294-307
15. Duffy MJ, Maguire TM, Hill A. et al. Metalloproteinases: role in breast carcinogenesis, invasion and metastasis. Breast Cancer Res. 2000;2:252-7
16. Kugler A. Matrix metalloproteinases and their inhibitors. Anticancer Res. 1999;19:1589-92
17. Jones JL, Glymp Walker RA. Expression of MMP-1 and MMP-9, their inhibitors, and activator MT1-MMP in primary breast carcinomas. J Pathol. 1999;189:161-8
18. Webber MM, Waghray A, Bello D. Prostate-specific antigen, a serine protease, facilitates human cancer cell invasion. Clin Cancer Res. 1995;1:1089-94
19. Stearns M, Stearns ME. Evidence for increased activated metalloproteinase 2 (MMP-2a) expression associated with human prostate cancer progression. Oncol Res. 1996;8:69-75
20. Abramjuk C, Jung K, Krell HW, Juchem P, Peters R, Taymoorian K. et al. Matrix metalloproteinase inhibitor RO 28-2653 in combination with estramustine: tumor-reduced effects on hormone-sensitive prostate cancer in rats. Anticancer Drug. 2005;16:855-61
21. Harvei S, Skijorten FJ, Robsahm TE, Berner A, Tretli S. Is prostaticibtraepithelial neoplasi in the transitio/central zone a true presursor of cancer? A long-tern retrospectivestudy in Norway. Br J Cancer. 1998;78:46-9
22. Goeman L, Joniau S, Ponette D, Van der Aa F, Roskams T, Oyen R, Van Poppel H. Is low-grade prostatic intraepithelial neoplasia a risk factor for cancer. Prostate Cancer Prostatic Dis. 2003;6:305-10
23. Page DL, Duppont WD, Rogers LW, Landenberger M. Intraductal carcinoma of the breast: follow up after biopsy only. Cancer. 1982;49:751-58
24. Page DL, Dupont WD, Rogers LW, Jensen RA, Schuyler PA. Continues local recurrence of carcinoma 15-25 years after a diagnosis of low grade ductal carcinoma in situ of the breast treated only by biopsy. Cancer. 1995;76:1197-2000
25. Eusebi V, Feudale E, Foschini MP. et al. Long-term follow-up of in situ carcinoma of the breast. Semin Diagn Pathol. 1994;11:223-35
26. Coussens LM, Fingleton B, Matrisian LM. Matrix metalloproteinase inhibitors and cancer: trial and tribulations. Science. 2002;295(5564):2387-92
27. Matrisian LM, Sledge GW Jr, Mohla S. Exacellular proteolysis and cancer: meeting summary and future directions. Cancer Res. 2003;63:6105-9
28. Malins DC, Anderson KM, Gilman NK, Green VM, Barker EA, Hellstrom KE. Development of a cancer DNA phenotype prior to tumor formation. Proc Natl Acad Sci USA. 2004;101:10721-5
29. Malins DC, Gilman NK, Green VM, Wheeler TM, Barker EA, Anderson KM. A DNA phenotype in healthy prostates, conserved in tumors and adjacent normal cells, implies a relationship to carcinogenesis. Proc Natl Acad Sci USA. 2005;102:19093-6
30. Pentheroudakis G, Briasoulis E, Pavlidis N. Cancer of unknown primary site: missing primary or missing biology? Oncologist. 2007;12(4):418-25
31. Boon T, Coulie PG, Van den Eynde B. Tumor antigens recognized by T cells. Immunol Today. 1997;18:267-268
32. Baxevanis CN, Dedoussis GV, Papadopoulos NG, Missitzis I, Stathopoulos GP, Papamichail M. Tumor specific cytolysis by tumor infiltrating lymphocytes in breast cancer. Cancer. 1994;74:1275-1282
33. van der Bruggen P, Traversari C, Chomez P, Lurquin C, De Plaen E, Van den Eynde BJ, Knuth A, Boon T. A gene encoding an antigen recognized by cytolytic T lymphocytes on a human melanoma. Science. 1991;254:1643-1647
34. Suzuki H, Chikazawa N, Tasaka T, Wada J, Yamasaki A, Kitaura Y, Sozaki M, Tanaka M, Onishi H, Morisaki T, Katano M. Intratumoral CD8(+) T/FOXP3 (+) cell ratio is a predictive marker for survival in patients with colorectal cancer. Cancer Immunol Immunother. 2010;59:653-661
35. Nosho K, Baba Y, Tanaka N, Shima K, Hayashi M, Meyerhardt JA, Giovannucci E, Dranoff G, Fuchs CS, Ogino S. Tumour-infiltrating T-cell subsets, molecular changes in colorectal cancer, and prognosis: cohort study and literature review. J Pathol. 2010;22:350-366
36. Pagès F, Berger A, Camus M, Sanchez-Cabo F, Costes A, Molidor R, Mlecnik B, Kirilovsky A, Nilsson M, Damotte D, Meatchi T, Bruneval P, Cugnenc PH, Trajanoski Z, Fridman WH, Galon J. Effector memory T cells, early metastasis, and survival in colorectal cancer. N Engl J Med. 2005;353:2654-2566
37. Gannot G, Gannot I, Vered H, Buchner A, Keisaris Y. Increase in immune cell infiltration with progression of oral epithelium from hyperkeratiosis to dysplasia and carcinoma. Br J Cancer. 2002;86:1444-1448
38. MacLennan GT, Eisenberg R, Fleshman RL, Taylor JM, Fu P, Resnick MI, Gupta S. The influence of chronic inflammation in prostatic carcinogenesis: a 5-year follow-up study. J Urol. 2006;176:1012-1016
39. Smith CJ, Gardner WA Jr. Inflammation-proliferation: possible relationships in the prostate. Prog Clin Biol Res. 1987;239:317-325
40. Topalian SL, Solomon D, Rosenberg SA. Tumor-specific cytolysis by lymphocytes infiltrating human melanomas. J Immunol. 1989;142(10):3714-25
41. Benson JL, Niederkorn JY. In situ suppression of delayed-type hypersensitivity: another mechanism for sustaining the immune privilege of the anterior chamber. Immunology. 1991Sep;74(1):153-9
42. Ioannides CG, Platsoucas CD, Rashed S, Wharton JT, Edwards CL, Freedman RS. Tumor cytolysis by lymphocytes infiltrating ovarian malignant ascites. Cancer Res. 1991Aug15;51(16):4257-65
43. Hald J, Rasmussen N, Claesson MH. Tumour-infiltrating lymphocytes mediate lysis of autologous squamous cell carcinomas of the head and neck. Cancer Immunol Immunother. 1995Oct;41(4):243-50
44. Radoja S, Frey AB. Cancer-induced defective cytotoxic T lymphocyte effector function: another mechanism how antigenic tumors escape immune-mediated killing. Mol Med. 2000Jun;6(6):465-79
45. Calzascia T, Di Berardino-Besson W, Wilmotte R, Masson F, de Tribolet N, Dietrich PY, Walker PRCutting edge. cross-presentation as a mechanism for efficient recruitment of tumor-specific CTL to the brain. J Immunol. 2003Sep1;171(5):2187-91
46. Santin AD, Bellone S, Palmieri M, Bossini B, Roman JJ, Cannon MJ, Bignotti E, Canè S. Pecorelli SInduction of tumor-specific cytotoxicity in tumor infiltrating lymphocytes by HPV16 and HPV18 E7-pulsed autologous dendritic cells in patients with cancer of the uterine cervix. Gynecol Oncol. 2003May;89(2):.271-80
47. Manning EA, Ullman JG, Leatherman JM, Asquith JM, Hansen TR, Armstrong TD, Hicklin DJ, Jaffee EM, Emens LA. A vascular endothelial growth factor receptor-2 inhibitor enhances antitumor immunity through an immune-based mechanism. Clin Cancer Res. 2007Jul1;13(13):3951-9
48. Hakim AA. A mechanism by which human breast carcinoma cells escape the host immune system. Neoplasma. 1988;35(6):691-705
49. O'Connell J, Bennett MW, O'Sullivan GC, Roche D, Kelly J, Collins JK, Shanahan F. Fas ligand expression in primary colon adenocarcinomas: evidence that the Fas counterattack is a prevalent mechanism of immune evasion in human colon cancer. J Pathol. 1998Nov;186(3):240-6
50. Bennett MW, O'connell J, O'sullivan GC, Roche D, Brady C, Kelly J, Collins JK, Shanahan F. Expression of Fas ligand by human gastric adenocarcinomas: a potential mechanism of immune escape in stomach cancer. Gut. 1999Feb;44(2):156-62
51. Nagao M, Nakajima Y, Kanehiro H, Hisanaga M, Aomatsu Y, Ko S, Tatekawa Y, Ikeda N, Kanokogi H, Urizono Y, Kobayashi T, Shibaji T, Kanamura T, Ogawa S, Nakano H. The impact of interferon gamma receptor expression on the mechanism of escape from host immune surveillance in hepatocellular carcinoma. Hepatology. 2000Sep;32(3):491-500
52. Ni X, Hazarika P, Zhang C, Talpur R, Duvic M. Fas ligand expression by neoplastic T lymphocytes mediates elimination of CD8+ cytotoxic T lymphocytes in mycosis fungoides: a potential mechanism of tumor immune escape? Clin Cancer Res. 2001Sep;7(9):2682-92
53. Nakamura Y, Yamazaki K, Oizumi S, Nakashima M, Watanabe T, Dosaka-Akita H, Nishimura M.Expression of RCAS1 in human gastric carcinoma. a potential mechanism of immune escape. Cancer Sci. 2004Mar;95(3):260-5
54. Ye Y, Zhou L, Xie X, Jiang G, Xie H, Zheng S. Interaction of B7-H1 on intrahepatic cholangiocarcinoma cells with PD-1 on tumor-infiltrating T cells as a mechanism of immune evasion. J Surg Oncol. 2009Nov1;100(6):500-4
55. Finke J, Ko J, Rini B, Rayman P, Ireland J, Cohen P. MDSC as a mechanism of tumor escape from sunitinib mediated anti-angiogenic therapy. Int Immunopharmacol. 2011Jul;11(7):856-61
56. Lança T, Silva-Santos B. The split nature of tumor-infiltrating leukocytes: Implications for cancer surveillance and immunotherapy. Oncoimmunology. 2012Aug1;1(5):717-725
57. Jochems C, Schlom J. Tumor-infiltrating immune cells and prognosis: the potential link between conventional cancer therapy and immunity. Exp Biol Med (Maywood). 2011May1;236(5):567-79
58. Pollard JW. Tumour-educated macrophages promote tumour progression and metastasis. Nat Rev Cancer. 2004;4:71-78
59. Wyckoff J, Wang W, Lin EY, Wang Y, Pixley F, Stanley ER, Graf T, Pollard JW, Segall J, Condeelis J. A paracrine loop between tumor cells and macrophages is required for tumor cell migration in mammary tumors. Cancer Res. 2004;64(19):7022-7029
60. Wyckoff JB, Wang Y, Lin EY, Li JF. Goswami S, Stanley ER, Segall JE, Pollard JW, Condeelis J. Direct visualization of macrophage-assisted tumor cell intravasation in mammary tumors. Cancer Res. 2007;67(6):2649-2656
61. Pollard JW. Macrophages define the invasive microenvironment in breast cancer. J Leukoc Biol. 2008Sep;84(3):623-30
62. Haley PJ. Species differences in the structure and function of the immune system. Toxicology. 2003;188:49-71
63. Tayeh MA, Marletta MA. Macrophage oxidation of L-arginine to nitric oxide, nitrite, and nitrate. Tetrahydrobiopterin is required as a cofactor. J Biol Chem. 1989;264:19654-19658
64. Schoedon G, Schneemann M, Hofer S, Guerrero L, Blau N, Schaffner A. Regulation of the L-arginine-dependent and tetrahydrobiopterin-dependent biosynthesis of nitric oxide in murine macrophages. Eur J Biochem. 1993;213:833-839
65. Schneemann M, Schoedon G. Species differences in macrophage NO production are important. Nat Immunol. 2002;3:102
66. Schneemann M, Schoeden G. Macrophage biology and immunology: man is not a mouse. J Leukoc Biol. 2007;81:579
67. Mestas J, Hughes CC. Of mice and not men: differences between mouse and human immunology. J Immunol. 2004;172:2731-2738
68. Pfister H, Remer KA, Brcic M, Fatzer R, Christen S, Leib S, Jungi TW. Inducible nitric oxide synthase and nitrotyrosine in listeric encephalitis: a cross-species study in ruminants. Vet Pathol. 2002;39:190-199
69. Perez LE, Chandrasekar B. et al. Reduced nitric oxide synthase 2 (NOS2) promoter activity in the Syrian hamster renders the animal functionally deficient in NOS2 activity and unable to control an intracellular pathogen. J Immunol. 2006;176:5519-5528
70. Doeing DC. et al. Crockett. Gender dimorphism in differential peripheral blood leukocyte counts in mice using cardiac, tail, foot, and saphenous vein puncture methods. BMC Clin. Pathol. 2003;3:3
71. Sitnicka E. et al. Jacobsen. Human CD34+ hematopoietic stem cells capable of multilineage engrafting NOD/SCID mice express flt3: distinct flt3 and c-kit expression and response patterns on mouse and candidate human hematopoietic stem cells. Blood. 2003;102:881
72. Ganz T. Defensins: antimicrobial peptides of innate immunity. Nat Rev Immunol. 2003;3:710
73. Ouellette AJ. et al. Paneth cell defensins: endogenous peptide components of intestinal host defense. FASEB J. 1996;10:1280
74. Risso A. Leukocyte antimicrobial peptides: multifunctional effector molecules of innate immunity. J Leukocyte Biol. 2000;68:785
75. Li L, Yan B, Shi YQ, Zhang WQ, Wen ZL. Live imaging reveals differing roles of macrophages and neutrophils during Zebrafish tail fin regeneration. J Biol Chem. 2012 [Epub ahead of print]
76. Cale CM. Cytokines and macrophages: implications for normal and abnormal renal development. Pediatr Nephrol. 1999;13(8):709-15
77. Shrestha A, Horino K, Nishiura H, Yamamoto T. Acquired immune response as a consequence of the macrophage-dependent apoptotic cell clearance and role of the monocyte chemotactic S19 ribosomal protein dimer in this connection. Lab Invest. 1999;79(12):1629-42
78. Barker RN, Erwig L, Pearce WP, Devine A, Rees AJ. Differential effects of necrotic or apoptotic cell uptake on antigen presentation by macrophages. Pathobiology. 1999;67(5-6):302-5
79. Abumaree MH, Chamley LW, Badri M, El-Muzaini MF. Trophoblast debris modulates the expression of immune proteins in macrophages: a key to maternal tolerance of the fetal allograft? J Reprod Immunol. 2012;94(2):131-41
80. Ting Gui, Aiko Shimokado, Yujing Sun, Takashi Akasaka, Yasuteru Muragaki. Diverse Roles of Macrophages in Atherosclerosis: From Inflammatory Biology to Biomarker discovery. Mediators Inflamm. 2012 doi: 10.1155/2012/693083
81. Salnikov AV, Liu L, Platen M, Gladkich J, Salnikova O, Ryschich E. et al. Hypoxia Induces EMT in Low and Highly Aggressive Pancreatic Tumor Cells but Only Cells with Cancer Stem Cell Characteristics Acquire Pronounced Migratory Potential. PLoS One. 2012;7(9):e46391
82. Waterman RS, Henkle SL, Betancourt AM. Mesenchymal Stem Cell 1 (MSC1)-Based Therapy Attenuates Tumor Growth Whereas MSC2-Treatment Promotes Tumor Growth and Metastasis. PLoS One. 2012;7(9):e45590
83. Siu MK, Wong ES, Kong DS, Chan HY, Jiang L, Wong OG. et al. Stem cell transcription factor NANOG controls cell migration and invasion via dysregulation of E-cadherin and FoxJ1 and contributes to adverse clinical outcome in ovarian cancers. Oncogene. 2012 doi: 10.1038/onc.2012.363
84. Zhou C, Liu J, Tang Y, Liang X. Inflammation linking EMT and cancer stem cells. Oral Oncol. 2012 [Epub ahead of print]
85. Hung SP, Yang MH, Tseng KF, Lee OK. Hypoxia-induced Secretion of TGF-beta 1 in Mesenchymal Stem Cell Promotes Breast Cancer Cell Progression. Cell Transplant. 2012 [Epub ahead of print]
86. DeNarddo DG, Johansson M, Coussens LM. Immune cells as mediators of solid tumor metastasis. Cancer Metastasis Rev. 2008;27(1):11-18
87. DeNardo DG, Andreu P, Coussens LM. Interactions between lymphocytes and myeloid cells regulate pro- versus anti-tumor immunity. Cancer Metastasis Rev. 2010Jun;29(2):309-16
88. Doedens AL, Stockmann C, Rubinstein MP, Liao D, Zhang N, DeNardo DG, Coussens LM, Karin M, Goldrath AW, Johnson RS. Macrophage expression of hypoxia-inducible factor-1 alpha suppresses T-cell function and promotes tumor progression. Cancer Res. 2010;70(19):7465-75
89. Erez N, Coussens LM. Leukocytes as paracrine regulators of metastasis and determinants of organ-specific colonization. Int J Cancer. 2011Jun1;128(11):2536-44 doi: 10.1002/ijc.26032. Epub 2011 Mar 25
90. Coussens LM, Pollard JW. Leukocytes in mammary development and cancer. Cold Spring Harb Perspect Biol. 2011 doi: 10.1101/cshperspect.a003285
91. Aichel O. About cell fusion with quantitatively abnormal chromosome distribution as cause for tumor formation. In: (ed.) Rowx W. Vortrageund Aufsatze Uber Entvicklungsmechanik der Organismen. Leipzig, Germany: Wilhelm Engelmann. 1911:92-111
92. Pawelek JM, Chakraborty AK. The cancer cell--leukocyte fusion theory of metastasis. Adv Cancer Res. 2008;101:397-444
93. Pawelek JM. Tumour-cell fusion as a source of myeloid traits in cancer. Lancet Oncol. 2005Dec;6(12):988-93
94. Pawelek JM. Viewing malignant melanoma cells as macrophage-tumor hybrids. Cell Adh Migr. 2007;1(1):2-6
95. Pawelek JM, Chakraborty AK. Fusion of tumour cells with bone marrow-derived cells: a unifying explanation for metastasis. Nat Rev Cancer. 2008May;8(5):377-86
96. Pawelek JM. Cancer-cell fusion with migratory bone-marrow-derived cells as an explanation for metastasis: new therapeutic paradigms. Future Oncol. 2008Aug;4(4):449-52
97. Lazova R, Chakraborty A, Pawelek JM. Leukocyte-cancer cell fusion: initiator of the warburg effect in malignancy? Adv Exp Med Biol. 2011;714:151-72
98. Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P. et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004;10:942-9
99. Facciabene A, Peng X, Hagemann IS, Balint K, Barchetti A, Wang LP. et al. Tumour hypoxia promotes tolerance and angiogenesis via CCL28 and T(reg) cells. Nature. 2011;475:226-30 doi: 10.1038/nature10169. [PubMed] [Cross Ref.]
100. Pagès F, Berger A, Camus M, Sanchez-Cabo F, Costes A, Molidor R. et al. Effector memory T cells, early metastasis, and survival in colorectal cancer. N Engl J Med. 2005;353:2654-66
101. Zhang L, Conejo-Garcia JR, Katsaros D, Gimotty PA, Massobrio M, Regnani G. et al. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N Engl J Med. 2003;348:203-13
102. Yu P, Lee Y, Liu W, Krausz T, Chong A, Schreiber H. et al. Intratumor depletion of CD4+ cells unmasks tumor immunogenicity leading to the rejection of late-stage tumors. J Exp Med. 2005;201:779-91
103. Buckner JH. Mechanisms of impaired regulation by CD4(+)CD25(+)FOXP3(+) regulatory T cells in human autoimmune diseases. Nat Rev Immunol. 2010;10:849-59
104. Singh A, Dey AB, Mohan A, Sharma PK, Mitra DK. Foxp3(+) Regulatory T Cells among Tuberculosis Patients: Impact on Prognosis and Restoration of Antigen Specific IFN-γ Producing T Cells. PLoS One. 2012;7(9):e44728
105. Jewett A, Tseng HC, Arasteh A, Saadat S, Christensen RE, Cacalano NA. Natural killer cells preferentially target cancer stem cells; role of monocytes in protection against NK cell mediated lysis of cancer stem cells. Curr Drug Deliv. 2012Jan;9(1):5-16
106. Bose A, Chakraborty T, Chakraborty K, Pal S, Baral R. Dysregulation in immune functions is reflected in tumor cell cytotoxicity by peripheral blood mononuclear cells from head and neck squamous cell carcinoma patients. Cancer Immun. 2008;8:10
107. Barkholt L, Alici E, Conrad R, Sutlu T, Gilljam M, Stellan B. et al. Safety analysis of exvivo-expanded NK and NK-like cells administered to cancer patients: a phase 1 clinical study. immunotherapy. 2009Sep;1(5):753-64
108. Szkaradkiewicz A, Karpiński TM, Drews M, Borejsza-Wysocki M, Majewski P. et al. Natural killer cell cytotoxicity and immunosuppressive cytokines (IL-10, TGF-beta1) in patients with cancer. J Biomed Biotechnol. 2010:901564
109. Shen Y, Lu C, Tian W, Wang L, Cui B, Jiao Y. et al. Possible association of decreased NKG2D expression levels and suppression of the activity of natural killer cells in patients with colorectal cancer. Int J Oncol. 2012;40(4):1285-90
110. Jewett A, Cacalano NA, Head C, Teruel A. Co-engagement of CD16 and CD94 receptors mediates secretion of chemokines and induces apoptotic death of naive natural killer cells. Clin Cancer Res. 2006;12(7 Pt 1):1994-2003
111. Jewett A, Teruel A, Romero M, Head C, Cacalano N. Rapid and potent induction of cell Death and loss of NK cell cytotoxicity against oral tumors by F(ab')2 fragment of anti-CD16 antibody. Cancer Immunol Immunother. 2008;57(7):1053-66
112. Sakakibara M, Kanto T, Hayakawa M, Kuroda S, Miyatake H, Itose I. et al. Comprehensive iimmunological analyses of colorectal cancer patients in the phase I/II study of quickly matured dendritic cell vaccine pulsed with carcinoembryonic antigen peptide. Cancer Immunol Immunother. 2011;60(11):1565-75
113. Sun Z, Shi L, Zhang H, Shao Y, Wang Y, Lin Y. et al. 2011. Immune modulation and safety profile of adoptive immunotherapy using expanded autologous activated lymphocytes against advanced cancer. Clin Immunol. 2011Jan;138(1):23-32
114. Wu X, Shao Y, Tao Y, Ai G, Wei R, Meng X. et al. Proteasome inhibitor lactacystin augments natural killer cell cytotoxicity of myeloma via downregulation of HLA class I. Biochem Biophys Res Commun. 2011Nov11;415(1):187-92
115. Tseng HC, Arasteh A, Paranjpe A, Teruel A, Yang W, Behel A. et al. Increased lysis of stem cells but not their differentiated cells by natural killer cells; de-differentiation or reprogramming activates NK cells. PLoS One. 2010Jul16;5(7):e11590
116. Jewett A, Tseng HC. Tumor induced inactivation of natural killer cell cytotoxic function; implication in growth, expansion and differentiation of cancer stem cells. J Cancer. 2011;2:443-57
117. Jewett A, Tseng HC, Arasteh A, Saadat S, Christensen RE, Cacalano NA. Natural killer cells preferentially target cancer stem cells; role of monocytes in protection against NK cell mediated lysis of cancer stem cells. Curr Drug Deliv. 2012Jan;9(1):5-16
118. Jewett A, Tseng HC. Potential rescue, survival and differentiation of cancer stem cells and primary non-transformed stem cells by monocyte-induced split anergy in natural killer cells. Cancer Immunol Immunother. 2012Feb;61(2):265-74
119. Yousefi M, Mattu R, Gao C, Man YG. Mammary ducts with and without focal myoepithelial cell layer disruptions show a different frequency of white blood cell infiltration and growth pattern: Implications for tumor progression and invasion. AIMM. 2005;13:30-37
120. Man YG, Shen T, Zhao YG, Sang QX. Focal prostate basal cell layer disruptions and leukocyte infiltration are correlated events: A potential mechanism for basal cell layer disruptions and tumor invasion. Cancer Detect Prev. 2005;29:161-169
121. Man YG. Focal degeneration of aged or injured myoepithelial cells and the resultant auto- immunoreactions are trigger factors for breast tumor invasion. Medical Hypotheses. 2007;69:1340- 1357
122. Man YG, Gardner WA. Focal degeneration of basal cells and the resultant auto-immunoreactions: a novel mechanism for prostate tumor progression and invasion. Med Hypoth. 2008;70:387-408
123. Hsiao YH, Deng CX, Mason JT, Chou MC, Man YG. Hidden malignant cells within leukocyte aggregates in normal or hyperplastic tissues of pregnancy-associated breast cancer. Cancer Epidemiol. 2011 [Epub ahead of print]
124. Liu AJ, Wei LX, Gardner WA, Man YG. Correlated alterations in prostate basal cell layer and basement membrane. Int J Biol Sci. 2009;5:276-285
125. Man YG, Tai L, Barner R, Vang R, Saenger JS, Shekitka KM, Bratthauer GL, Wheeler DT, Liang CL, Vinh TN, Strauss BL. Cell clusters overlying focally disrupted mammary myoepithelial cell layers and adjacent cells within the same duct display different immunohistochemical and genetic features: implications for tumor progression and invasion. Breast Cancer Res. 2003;5:231-241
126. Man YG, Zhang Y, Shen T, Vinh TN, Zeng X, Tauler J, Mulshine JL, Strauss BL. cDNA expression profiling identifies elevated expressions of tumor progression and invasion related genes in cell clusters of in situ breast tumors. Breast Cancer Res Treat. 2005;89:199-208
127. Man YG, Zhao CQ, Wang J. Breast tumor cell clusters and their budding derivatives show different immunohistochemical profiles during stromal invasion: implications for hormonal and drug therapies. Cancer Therapy. 2006;4:193-204
128. Zhang XC, Hashemi SS, Yousefi M, Gao CL, Sheng J, Mason J, Man YG. Atypical expression of c-erbB2 in cell clusters overlying focally disrupted breast myoepithelial cell layers: a potential sign for increasing cell motility and invasion. Int J Biol Sci. 2008;4:259-269
129. Man YG. Tumor cell budding from focally disrupted tumor capsules: a common pathway for all breast cancer subtype derived invasion? Journal of Cancer. 2010;1:27-31
130. Man YG. A seemingly most effective target for early detection and intervention of prostate tumor invasion. J Cancer. 2010;1:63-69
131. Song GH, Ren J, Stojadinovic A, Yong HY, Chen W, Liu ML, Grinkemeyer M, Man YG. Differential impact of lymphocyte infiltration on basal and luminal cells: Implications for breast and prostate tumor invasion and metastasis. Submitted.
132. Bonkhoff H, Stein U, Remberger K. The proliferative function of basal cells in the normal and hyperplastic human prostate. Prostate. 1994;24:114-8
133. Bounkhoff H. Role of the basal cells in premalignant changes of the human prostate: a stem cell concept for the development of prostate cancer. Eur Urol. 1996;30:201-5
134. Botticelli AR. A theoretical rationale on the histogenesis of premalignant lesions and early carcinoma of the prostate. Pathologica. 1994;86:128-41
135. Zou Z, Anisowicz A, Hendrix MJ, Thor A, Neveu M, Sheng S, Rafidi K, Seftor E, Sager R. Maspin, a serpin with tumor-suppressing activity in human mammary epithelial cells. Science. 1994;263:526-29
136. Sheng S, Pemberton PA, Sager R. Production, purification, and characterization of recombinant maspin proteins. J Biol Chem. 1994;269:30988-93
137. Barbareschi M, Pecciarini L, Cangi MG. et al. p63, a p53 homologue, is a selective nuclear marker of myoepithelial cells of the human breast. Am J Surg Pathol. 2001;25:1954-60
138. Sternlight MD, Barsky SH. The myoepithelial defense: a host defense against cancer. Med Hypotheses. 1997;48:37- 46
139. Brummer T, Schramek D, Hayes VM, Bennett HL, Callon CE, Musgrove EA, Daly RJ. Increased proliferation and altered growth factor dependence of human mammary epithelial cells overexpressing the Gab2 docking protein. J Biol Chem. 2006;281:626-31
140. Oliveira AM, Ross JS, Fletcher JA. Tumor suppressor genes in breast cancer: the gatekeepers and the caretakers. Am J Clin Pathol. 2005;124(Suppl):S16-28
141. Yanochko GM, Eckhart W. Type 1 insulin-like growth factor overexpression induce proliferation and anti-apoptotic signaling in a three-dimensional culture model of breast epithelial cells. Breast Cancer Res. 2006;8(2):R18-R23
142. Boulikas T. Control of DNA replication by protein phosphorylation. Anticancer Res. 1994;14:2465-72
143. Boulikas T. Phosphorylation of transcription factors and control of the cell cycle. Crit Rev Eukaryot Gene Expr. 1995;5:1-77
144. Chakravarthy MV, Spangenhurg EE, Booth FW. Culture in low levels of oxygen enhances in vitro proliferation potential of satellite cells from old skeletal muscles. Cell Mol Life Sci. 2001;58:1150-58
145. Csete M, Walikonis J, Slawny N. et al. Oxygen-mediated regulation of skeletal muscle satellite cell proliferation and adipogrnesis in culture. J Cell Physical. 2001;189:189-96
146. Studer L, Csete M, Lee SH. Enhanced proliferation, survival, and dopaminergic differentiation of CNS precursors in lowed pxygen. J Neurosci. 2000;20:7377-83
147. Asano-kato N, Fukagawa K, Okada N. Tryptase increase proliferative activity of human conjunctive fibroblasts through protease-activated receptor. Invest Ophthalmol Vis Sci. 2005;46:4622-6
148. Freeman MR, Schneck FX, Gognon ML, Corless C, Soker S, Niknejad K. et al. Peripheral blood T- lymphocytes and lymphocytes infiltrating human cancers express vascular endothelianl growth factor: a potential role for T cells in angiogenesis. Cancer Res. 1995;55:4140-5
149. Nienartowicz A, Sobaniee-Lotowska ME. et al. Mast cells in neoangiogenesis. Med Sci Monit. 2006;12(3):53-6
150. Qu Z. Immunohistological detection of growth factors and cytokines in tissue mast cells. Methods Mol Biol. 2006;315:257-72
151. Takahashi Y, Bucana CD, Liu W. et al. Platelet-derived endothelial cell growth factor in human colon cancer angiogenesis: role of infiltrating cells. J Natl Cancer Inst. 1996;88:1146-51
152. Kang Y, Massague J. Epithelial-mesenchymal transition: twist in development and metastasis. Cell. 2004;118:277-9
153. Sato T, Sakai T, Noguchi Y, Takita M, Hirakawa S, Ito A. Tumor-stromal cell contact promotes invasion of human uterine cervical carcinoma cells by augmenting the expression and activation of stromal matrix metalloproteinases. Gynecol Oncol. 2004;92:47-56
154. Strizzi L, Bianco C, Normanno N. Epithelial mesenchymal transition is a characteristic of hyperplasias and tumors in mammary gland from MMTV-Criptol-1 transgenic mice. J Cell Physiol. 2004;201:266-76
155. Schedin P. Pregnancy-associated breast cancer and metastasis. Nat Rev Cancer. 2006Apr;6(4):281-91
156. Hu M, Polyak K. Microenvironmental regulation of cancer development. Curr Opin Genet Dev. 2008;18(1):27-34
157. Puspa Raj Pandey PR, Saidou J, Watabe K. Role of myoepithelial cells in breast tumor progression. Front Biosci. 2011;15:226-236
158. Man YG, Harley R, Mason J, Gardner WA. Contributions of leukocytes to tumor invasion and metastasis: the “piggy-back” hypothesis. Cancer Epidem. 2010;34:3-6
159. Man YG, Mason J, Harley R, Kim YH, Zhu KM, Gardner WA. Leukocyte-facilitated tumor dissemination: a novel model for tumor cell dissociation and metastasis. J Cell Biochem. 2011;112:1154-1167
160. Man YG. Aberrant leukocyte infiltration: a direct trigger for breast tumor invasion and metastasis. Int J Biol Sci. 2010;6(2):129-132
161. Waterhouse NJ, Clarke CJ, Sedelies KA, Teng MW, Trapani JA. Cytotoxic lymphocytes; instigators of dramatic target cell death. Biochem Pharmacol. 2004;68(6):1033-40
162. Cullen SP, Brunet M, Martin SJ. Granzymes in cancer and immunity. Cell Death Differ. 2010;17(4):616-623
163. Topham NJ, Hewitt EW. Natural killer cell cytotoxicity: how do they pull the trigger? Immunology. 2009;128(1):7-15
164. de Saint Basile G, Ménasché G, Fischer A. Molecular mechanisms of biogenesis and exocytosis of cytotoxic granules. Nat Rev Immunol. 2010;10(8):568-579
165. Alga AM, Rothwell PM. Effects of regular asprin on long-term cancer incidence and metastasis: a systematic comparison of evidence from observational studies versus randomised trials. Lancet Oncol. 2012 Epub ahead of print
Author contact
![]() Corresponding author: Yan-gao Man., MD., PhD., Senior scientist, Diagnostic and Translational Research Center, Henry Jackson Foundation, MD, USA. Phone: 240-833-4969; E-mail: 240-833-4940; E-mail: ymannorg. Or: Anahid Jewett, PhD, MPH, Professor and Director of Tumor Immunology Laboratory, Jonsson Comprehensive Cancer Center, UCLA School of Dentistry and Medicine, Los Angeles, CA, USA. Phone: 310-206-1970; E-mail: ajewettucla.edu.
Corresponding author: Yan-gao Man., MD., PhD., Senior scientist, Diagnostic and Translational Research Center, Henry Jackson Foundation, MD, USA. Phone: 240-833-4969; E-mail: 240-833-4940; E-mail: ymannorg. Or: Anahid Jewett, PhD, MPH, Professor and Director of Tumor Immunology Laboratory, Jonsson Comprehensive Cancer Center, UCLA School of Dentistry and Medicine, Los Angeles, CA, USA. Phone: 310-206-1970; E-mail: ajewettucla.edu.
Citation styles
APA
Man, Y.g., Stojadinovic, A., Mason, J., Avital, I., Bilchik, A., Bruecher, B., Protic, M., Nissan, A., Izadjoo, M., Zhang, X., Jewett, A. (2013). Tumor-Infiltrating Immune Cells Promoting Tumor Invasion and Metastasis: Existing Theories. Journal of Cancer, 4(1), 84-95. https://doi.org/10.7150/jca.5482.
ACS
Man, Y.g.; Stojadinovic, A.; Mason, J.; Avital, I.; Bilchik, A.; Bruecher, B.; Protic, M.; Nissan, A.; Izadjoo, M.; Zhang, X.; Jewett, A. Tumor-Infiltrating Immune Cells Promoting Tumor Invasion and Metastasis: Existing Theories. J. Cancer 2013, 4 (1), 84-95. DOI: 10.7150/jca.5482.
NLM
Man Yg, Stojadinovic A, Mason J, Avital I, Bilchik A, Bruecher B, Protic M, Nissan A, Izadjoo M, Zhang X, Jewett A. Tumor-Infiltrating Immune Cells Promoting Tumor Invasion and Metastasis: Existing Theories. J Cancer 2013; 4(1):84-95. doi:10.7150/jca.5482. https://www.jcancer.org/v04p0084.htm
CSE
Man Yg, Stojadinovic A, Mason J, Avital I, Bilchik A, Bruecher B, Protic M, Nissan A, Izadjoo M, Zhang X, Jewett A. 2013. Tumor-Infiltrating Immune Cells Promoting Tumor Invasion and Metastasis: Existing Theories. J Cancer. 4(1):84-95.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) License. See http://ivyspring.com/terms for full terms and conditions.